
Abstract
Retrotransposons are ubiquitous in the plant kingdom and constitute a large fraction of many plant genomes. Although most retrotransposons from plants were thought to be transcriptionally silent in somatic tissues, evidence of activity under certain conditions is available in some cases. In this study, a complete LTR retrotransposon was isolated from the cultivated strawberry (Fragaria × ananassa) genome using genome walking. The element, named FaRE1, has all the features of a typical Ty1-copia retrotransposon. Its total length was 5,104 bp, comprising a single 3,891 bp open reading frame. It is represented by ~96 copies per genome, equivalent to ~0.33% of the genome. Transcription of FaRE1 was detected in leaf tissue treated with various phytohormones, such as naphthalene acetic acid, 2,4-dichlorophenoxyacetic acid or abscisic acid . To our knowledge, this is the first report of the isolation of a complete LTR retrotransposon with transcriptional activity in strawberry.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Transposable elements (TEs) are mobile DNA sequences that are a common constituent of every genome studied to date. Those which transpose via a RNA intermediate are termed Class I elements or retrotransposons, while those in which transposition occurs directly are termed Class II elements or DNA transposons (Flavell et al. 1994). Retrotransposons are closely related to retroviruses with respect to both their structure and life cycle (Boeke and Corces 1989). They have been classified on the basis of whether or not they possess a long terminal repeat (LTR) sequence (Kumar and Bennetzen 1999). Among the LTR retrotransposons, two types (Ty1-copia and Ty3-gypsy) have been distinguished, mainly on the basis of the linear arrangement of their coding domains. Retrotransposons are ubiquitous in the plant kingdom and constitute a large fraction of many plant genomes, ranging from ~5.5% of the Arabidopsis thaliana genome to >50% of that of Zea mays (Hirochika 1993; Pearce et al. 1996; SanMiguel et al. 1996).
Most retrotransposons are believed to be either transcriptionally fully inactive (Kumar and Bennetzen 1999), or active only during particular stages of development or in response to stress (Hirochika 1993), wounding or pathogen infection (Kimura et al. 2001). Transcription of tobacco Tto1, Tnt1, Tnp2, rice Tos17, oat OARE-1 and orange CIRE1 has been demonstrated (Grandbastien et al. 1989; Vaucheret et al. 1992; Hirochika 1993; Hirochika et al. 1996; Kimura et al. 2001; Rico-Cabanas and Martínez-Izquierdo 2007) but, in all cases, transcriptional activity is limited to either stressful conditions or occurs only in a restricted number of tissues (Grandbastien 1998). Based on their abundance in all plant genomes, their stability during transposition, and their sensitivity to stress, retrotransposons are believed to play a number of significant biological roles (White et al. 1994; Kato et al. 1999; Lonnig and Saedler 2002).
The strawberry (Fragaria spp.) is an herbaceous perennial that propagates vegetatively. Micropropagated strawberry plants—a process which includes a period in tissue culture—are used widely in production. In many plant species—including strawberry (Nehra et al. 1991)—regenerated plants arising from tissue culture are phenotypically variable, an effect termed somaclonal variation. One of the mechanisms proposed to underlie somaclonal variation is retrotransposon activation (Tahara et al. 2004). Segments of both the Ty1-copia and Ty3-gypsy genes encoding reverse transcriptase (RT) were amplified from the genomic DNA of strawberry by Ma et al. (2008), who concluded that the contribution of the Ty1-copia type was greater than that of the Ty3-gypsy type. Here, we report the isolation of a complete, transcriptionally active, Ty1-copia retrotransposon (FaRE1) from the cultivated strawberry genome using genome walking, and study the extent to which FaRE1 expression is affected by the exogenous application of various phytohormones.
Materials and methods
Plant material
The strawberry cultivar “Allstar” was used throughout these experiments. To analyze the effect of phytohormones on retrotransposon expression, folded young leaves (~1.5 cm long) of greenhouse-cultivated plants were sprayed with 50 mM naphthalene acetic acid (NAA), 50 mM 2,4-dichlorophenoxyacetic acid (2,4-D) or 50 mM abscisic acid (ABA).
Nucleic acid extraction
Genomic DNA was extracted from fresh leaves following a modified CTAB method (Ma et al. 2008). Total RNA was isolated using the modified CTAB method, as described by Chang et al. (2007). All RNA samples were treated with DNase I (TaKaRa, Japan) at 37°C for 4 h before precipitating RNA. The concentration of nucleic acid in each preparation was estimated spectrophotometrically.
Genome walking
Isolation of the complete strawberry Ty1-copia retrotransposon was based on xp5-2, a 260 bp RT fragment isolated from strawberry leaves by RT-PCR (Ma 2008). The Universal Genome Walker kit (Clontech, Palo Alto, CA) was used to isolate the complete Ty1-copia retrotransposon sequence. Genomic DNA was blunt-end digested by DraI, EcoRV, PvuII or StuI (TaKaRa, Japan). The construction of Genome Walker libraries, and the purification and ligation of adaptors to restriction fragments were performed as described in the Universal Genome Walker kit manual. Amplification reactions were performed in a 20 μL volume based on the Advantage Genomic PCR Kit (Clontech), according to the manufacturer’s instructions. The first PCR consisted of 7 cycles of 94°C/10 s and 70°C/3 min, followed by 32 cycles of 94°C/10 s and 65°C/3 min. The second PCR consisted of 5 cycles of 94°C/10 s, and 70°C/3 min, followed by 20 cycles of 94°C/10 s, and 65°C/3 min. At the end of each PCR, an elongation step of 65°C/10 min was included.
Long-range PCR
Amplification was performed in 20 μL reactions containing 50 ng genomic DNA, 1 μM of each primer (see Table 1) targeted outside the FaRE1 sequence, 0.5 U LA Taq DNA polymerase in a reaction buffer that included 0.2 mM dNTP (TaKaRa, Japan). The cycling conditions consisted of an initial denaturing step of 94°C/3 min, followed by 35 cycles of 94°C/1 min, 55°C/1 min and 72°C/5 min, and ending with an elongation step of 72°C/10 min.
Dot blotting
TE copy number was estimated by hybridization between genomic DNA and a probe amplified from the strawberry genome in a 25 μL PCR comprising 25 ng genomic DNA, 200 μM dNTP, 1 μM of each primer directed at various TE domains (primer sequences listed in Table 1), and 1 U Ex Taq polymerase (TaKaRa, Japan). The amplification profile comprised an initial denaturation step of 94°C/3 min, followed by 35 cycles of 94°C/1 min, 55°C/1 min and 72°C/1 min, and a final elongation step of 72°C/10 min.
The amplified products were purified from 1.5% agarose gels, and labelled with digoxigenin-dUTP using the DIG High Prime DNA Labeling and Detection Starter Kit II (Roche, Mannheim, Germany), following the manufacturer’s instructions. Genomic DNA and the PCR products were denatured in 1 M NaOH, 0.2 M EDTA (pH 8.2) for 30 min, held at 100°C for 10 min, then quickly chilled. The denatured DNA was spotted onto an Immobilon-Ny+ membrane (Millipore, Bedford, MA) in various amounts (genomic DNA: 50, 100, 200, and 400 ng; PCR products: 25, 50, 100, and 200 pg). The membranes were incubated in DIG Easy hybridization buffer for 16 h at 62°C. After washing, the signal was visualized immunologically following the DIG High Prime DNA Labeling and Detection Starter Kit II protocol. A linear regression between the natural logarithm of probe sequence copy number and signal strength was used to infer copy number in the genome.
RT-PCR and northern blotting
cDNA was synthesized from total RNA using Reverse Transcriptase XL (AMV; TaKaRa) primed with dT18 and a random nonamer primer, following the manufacturer’s instructions. Amplification of cDNA was performed in a 20 μL reaction containing 1 μL cDNA, 1 μM of each primer (gag domain, integrase domain, reverse transcriptase domain, rnaseH domain—see Table 1), 0.5 U Taq DNA polymerase and buffer that included 0.2 mM dNTP (TaKaRa). The cycling conditions were an initial denaturation step (94°C/3 min), followed by 35 cycles of 94°C/1 min, 55°C/1 min and 72°C/1 min, and a final elongation step (72°C/10 min). Strawberry 18S ribosomal RNA was used as an internal control gene (Simovic et al. 1992).
For northern analysis, RNA (10 μg) was separated on a 1.2% formaldehyde agarose gel and then transferred to an Immobilon-Ny+ membrane (Millipore). The membrane was prehybridized and hybridized in DIG Easy hybridization buffer for 16 h at 42°C using reverse transcriptase domain cDNA probe labelled with digoxigenin-dUTP, as described in dot blotting.
DNA sequence analysis
PCR products were purified and recovered using the Agarose Gel DNA Purification and Recovered Kit (TaKaRa), ligated into pBS-T (Tiangen, China) and transformed into Escherichia coli strain JM109. DNA sequencing reactions employed M13 primers, and was performed by Shanghai Biological Engineering and Technology and Service (Shanghai, China). Each clone sequence was subjected to either BLASTN or BLASTX analysis (http://www.ncbi.nlm.nih.gov) and aligned using ClustalX (Thompson et al. 1997). These alignments were then used to generate neighbour-joining trees using MEGA4.1 (Kumar et al. 2008) with a 1,000 replicate bootstrap test. PlantCARE (http://www.oberon.rug.ac.be.8443/carebin/CallMat_IE55.htpl) and PLACE (http://www.dna.affrg.go.jp/htdocs/PLACE) databases were used for the analysis of the FaRE1 retrotransposon 5′-LTR domain.
Results
FaRE1, a novel Ty1-copia retrotransposon from strawberry
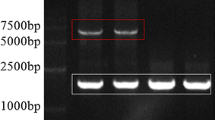
To isolate the complete sequence of Ty1-copia retrotransposons from F. × ananassa, a Ty1-copia retrotransposon RT sequence of 260 bp, named xp5-2, was used as an original sequences to commence genome walking. The sequence of xp5-2 was isolated from strawberry leaves treated with 50 mM ABA by RT-PCR with degenerate primers corresponding to reverse transcriptase domains I and III of Ty1-copia retrotransposons (Ma 2008). Two successive downstream and two successive upstream steps generated fragments that could be assembled into a 6,066 bp contig, which contained the complete LTR retrotransposon sequence with two almost perfect LTRs (490 bp), starting with TG and ending with CA. To ensure that the contig was not a mosaic of independent sequences, a primer pair was targeted outside the retrotransposon sequence, and used to amplify a ~5,200 bp fragment from the strawberry genome (Fig. 1). This sequence shared nearly 99% identity with the contig sequence. We have named the complete LTR retrotransposon sequence FaRE1 (Fragaria × ananassa retrotransposon), and deposited this sequence in GenBank (accession number FJ871121).

Long-range polymerase chain reaction (PCR) of the genomic region including FaRE 1. 1% agarose gel electrophoresis pattern of PCR amplification products using a pair of outside FaRE1-specific primers M 500 bp ladder marker
FaRE1 is 5,104 bp in length, and consists of a single 3,891 bp open reading frame (ORF). Its structure is typical of the Ty1-copia type, containing a pair of LTRs (490 bp) starting with TG and ending with CA. Each LTR has the 5 bp direct repeat 5′-CAAAT-3′ flanking the element (Fig. 2a), which probably represents a duplication of the genomic target site produced by transposon insertion (Konieczny et al. 1991). Both LTRs start and end with the 4 bp inverted repeat 5′-TGGT…ACCA-3′, including the retroviral consensus 5′-TG…CA-3′, which are thought to be important for integration (Liubomirskaia et al. 2003). The alignment of FaRE1 with other plant retrotransposons (Fig. 2a) showed that the ORF includes the genes encoding group-specific antigen (GAG), protease (PR), integrase (IN), reverse transcriptase (RT) and RNaseH (RH), ordered as in all Ty1-copia retrotransposons. A particularly high degree of identity was obtained between the peptide sequences encoded by FaRE1 and CIRE1 of orange, namely 30, 35, 43, 58, and 50 for, respectively, GAG, PR, IN, RT and RH. On the basis of peptide sequence, FaRE1 belongs within the class of Ty1-copia retrotransposons (Fig. 2b).

FaRE1, a novel Ty1-copia retrotransposon isolated from strawberry. a The structure of the FaRE1 retrotransposon is schematically shown at the top of the figure. Alignment of amino acid sequences of relevant domains corresponding to Gag, protease, integrase, reverse transcriptase and RNase H proteins of BARE1 of barley (Manninen and Schulman 1993), CIRE1 of sweet orange (Rico-Cabanas and Martínez-Izquierdo 2007), copia of Drosophila (Mount and Rubin 1985), Tlc1 of tomato (Yañez et al. 1998), Tnt1 of tobacco (Grandbastien et al. 1989) and Tto1 of tobacco (Hirochika 1993) retrotransposons are shown together with those encoded by the sequenced FaRE1 retrotransposon. b Dendrogram tree of conceptual translated amino acid sequences in the whole gag-pol region of FaRE1 and other retrotransposons. The tree was obtained using the Neighbor-joining method (Saitou and Nei 1987). Horizontal distances are proportional to evolutionary distances according to the scale shown on the bottom. The tree was displayed with the MEGA4.1 program showing bootstrap values (from 1,000 replicates) higher than 50%. In addition to the retrotransposons described in a, the amino acid sequences of Gypsy of Drosophila (Marlor et al. 1986), Maggy of Magnaporthe grisea (Farman et al. 1996), the fungal Skippy element (Anaya and Roncero 1995), Ted of baculovirus (Friesen and Nissen 1990) and Ty3 of yeast (Hansen et al. 1988) are also included in the comparison
A further feature typical of retroviruses and retrotransposons that is also present in FaRE1 is the primer binding site (PBS) adjacent to the 5′LTR. The FaRE1 PBS is 20 bp long and represents a potential tRNA-Met primer binding site for the initiation of minus strand synthesis (Fig. 2a). Typically, this sequence begins with TGG and is complementary to the 3′-end of the host-encoded tRNAs-Met that is used to prime minus strand DNA synthesis (Sprinzl et al. 1996). Upstream of the 3′LTR, FaRE1 has a 15 bp polypurine tract (Fig. 2a), preceded by seven pyrimidines characteristic of the retroid family putative priming site for plus strand DNA synthesis (Perlman and Boeke 2004). The positions of both priming sequences coincided with those described in other plant retrotransposons (Kumar and Bennetzen 1999).
FaRE1 copy number
The dot blot hybridization assay indicated that 200 ng genomic DNA gave the same strength of signal as 50 pg the LTR probe (Fig. 3). F. × ananassa has a 1C DNA value of 0.61 pg (598 Mb; Nehra et al. 1991) but, as it is an octoploid species, whose haploid consists of four genomes, the size of the strawberry genome is 149.5 Mb. Based on MacRae (1998), the FaRE1 copy number was calculated by multiplying the genome size by the average proportion of nuclear genomic DNA hybridizing to the probe, and dividing by the molecular weight of the probe sequence. On the basis of the LTR probe, this produced an estimate of 196 for the copy number of the LTR sequence. Applying the same calculation to GAG, IN, RT and RH resulted in estimates of, respectively 102, 101, 96 and 95 (Table 2). Since the ratio between the LTR and the other sequence copy numbers is effectively 2, it is clear that most, if not all, of the FaRE1 elements consist of a single ORF and two LTRs, which is characteristic of retrotransposons. RT corresponds to regions encoding enzymes critical to the retrotransposon life cycle and would be expected to be well conserved among retrotransposons. Therefore, the copy number of the FaRE1 retrotransposon was estimated using the RT probe, resulting in an estimated ~96 copies per genome. Based on a copy number of 96/genome, the proportion of the strawberry genome occupied by FaRE1 is ~0.33%. As the number of Ty1-copia retrotransposons in the genome is of the order of 2,875 (Ma et al. 2008), FaRE1 therefore represents around 3% of the population of Ty1-copia retrotransposons in the strawberry genome.

Dot blots used to estimate FaRE1 retrotransposon copy number in strawberry. Dots (LTR amplicons): 1 25 pg, 2 50 pg, 3 100 pg, 4 200 pg PCR product; Dots (genomic DNA): 1 50 ng, 2 100 ng, 3 200 ng, 4 400 ng
Transcriptional analysis of FaRE1
A search for eukaryotic promoter motifs upstream of the FaRE1 5′LTR was carried out by scanning the PlantCARE and PLACE databases. This led to the identification of 14 possible TATA boxes and various other DNA motifs associated with the plant response to endogenous or environmental factors. The effect on FaRE1 transcription level of the application of the phytohormones NAA, 2, 4-D or ABA to the leaf was investigated by RT-PCR and northern blotting. cDNA was generated from mRNA harvested 16 h after treatment, and used as a template for the amplification of the FaRE1 domains GAG, IN, RT and RH. RT-PCR found that the FaRE1 sequence was represented in the population of mRNA, meaning that the retrotransposon was transcribed under the specific conditions applied. In contrast, no band was observed in mRNA from leaves that had not been treated with hormone (Fig. 4). The result of northern blotting analysis also indicated that transcription of FaRE1 was induced by treatment with phytohormones (Fig. 5). Comparing to the sizes of 28S, 18S rRNA, the size of FaRE1 transcripts was estimated to be ~4.8 kb.

RT-PCR demonstrates transcriptional activity of FaRE1. a–d Amplifications of the FaRE1 domains: a GAG, b IN, c RT, d RH; e amplification of 18S internal control gene. Lanes: 1 Untreated leaves; 2 leaves incubated with naphthalene acetic acid (NAA); 3 leaves incubated with 2,4-dichlorophenoxyacetic acid (2,4-D); 4 leaves incubated with abscisic acid (ABA); 5 water control; 6–9 lanes contain non-reverse transcribed RNA as the PCR template; M 100 bp ladder marker

Northern blot analysis of the transcriptional activity of FaRE1. Lanes: 1 Untreated leaves; 2 leaves incubated with NAA; 3 leaves incubated with 2,4-D; 4 leaves incubated with ABA
Discussion
LTR retrotransposons are present in many plant genomes in very high copy numbers, but the majority of them are probably inactive. Transcriptionally active LTR retrotransposons are important because of the role they play in gene and genome evolution. In contrast to this agronomic importance, very little information about the strawberry genome has been reported. Recently, Pontaroli et al. (2009) identified 13 Ty1-copia retrotransposons, 14 Ty3-gypsy retrotransposons and 3 unclassified elements from a Fosmid library of wild diploid strawberry F. vesca subsp. americana; however, their transcriptional activity was unknown, and none of them showed regions of homology to known nuclear genes. Here, we have isolated FaRE1 from the cultivated strawberry genome, and shown that it is transcriptionally active, at least following phytohormone treatment. The FaRE1 retrotransposon has all the features of a Ty1-copia retrotransposon, including the predicted transcriptional and regulatory signals and conserved protein-coding domains. Its copy number was ~96/genome, which is equivalent to ~0.33% of the DNA present. The majority of the FaRE1 elements carry two LTRs per internal domain, similar to Grande in the maize genome (García-Martínez and Martínez-Izquierdo 2003) and CIRE1 in the orange genome (Rico-Cabanas and Martínez-Izquierdo 2007). We believe that this is the first report of a transcriptionally active LTR retrotransposon in Fragaria spp.
It is known that certain retrotransposons are activated by stress, and others by exogenously supplied phytohormones. Thus, for example, transcription of the tobacco element Tnt1A is induced by the provision of jasmonic acid (JA), and Tnt1C by salicylic acid and auxin (Beguiristain et al. 2001). Similarly, ABA and JA enhance the transcription of the barley element Bare1 and tobacco element Tto1 (Suoniemi et al. 1996; Takeda et al. 1998), while UV stress activates LTR retrotransposons in both Xenopus sp. (Shim et al. 2000) and oat (Kimura et al. 2001). During periods of high transpositional activity, some retrotransposon sequences are amplified to produce multi-copy, relatively homogeneous elements which then diverge, either by subsequent retrotransposition or by natural acquisition of mutations. It was estimated that actively transposing retrotransposons incorporate mutations at a rate approximately a million-fold higher than that of nuclear genes (Gojobori et al. 1990). Here, we have shown that FaRE1 transcription occurs in leaves treated with NAA, 2, 4-D or ABA. During tissue culture, strawberry plantlets grow in medium supplemented with lower concentrations of hormones. We are now are developing sequence-specific amplification polymorphism (S-SAP) markers based on the LTR sequences of FaRE1 to analyze whether FaRE1 is activated and transposes when strawberry plants are micropropagated, especially in medium supplemented with NAA and/or 2,4-D. This will allow us to elucidate whether transposition of retrotransposons could be one of the main reasons for somaclonal variation in micropropagated strawberry plants.
LTRs are important for the initiation of transcription and retrotransposition. Transcription of retroviruses and retrotransposons starts at the 5′LTR (Kumar and Bennetzen 1999) since the 5′LTR represents the promoter, which directs transcription by host cell RNA polymerase II. A characteristic of active plant retrotransposons is that they contain cis-regulatory elements in the U3 region of their 5′LTR, generally associated with signal transduction pathways related to the plant’s defense response. The 5′LTR of the Bare1 retrotransposon contains ABRE elements that respond to ABA (Suoniemi et al. 1996), while a 13-bp motif associated with Tto1 has been identified as a cis-regulatory sequence required to drive its induction by JA (Takeda et al. 1999). The TLC1.1 ethylene-responsive element in Lycopersicon chilense carries the 8 bp motif ATTTCAAA, which acts as an ethylene-responsive element box in the promoter region (Tapia et al. 2005). The FaRE1 LTR sequence includes some well-characterized promoter motifs, such as the TGTCTC box, the ACTTTA and GAGAC cores, and the YTGTCWC, WAACCA, CANNTG and AWTTCAAA motifs. The TGTCTC box (Hagen and Guilfoyle 2002; Goda et al. 2004; Nemhauser et al. 2004), ACTTTA (Baumann et al. 1999) and GAGAC (Maruyama-Nakashita et al. 2005) cores, and the YTGTCWC motif (Boyle and Brisson 2001) have all been associated with the response to auxin; while the WAACCA (Abe et al. 2003) and CANNTG (Abe et al. 2003) motifs respond to ABA. The AWTTCAAA motif is ethylene responsive (Tapia et al. 2005). We suggest that the FaRE1 5′LTR contains a functional promoter, and so are presently involved in constructing a FaRE1 LTR:GUS fusion for use as functional tool in transgenic strawberry.
In conclusion, this is the first description of a complete active Ty1-copia retrotransposon in strawberry. We have shown that FaRE1 expression occurs in leaves supplied with exogenous phytohormones. FaRE1 may play a role in gene and genome evolution in the cultivated strawberry.
Abbreviations
- cDNA:
-
Complementary deoxyribonucleic acid
- LTR:
-
Long terminal repeat
- ORF:
-
Open reading frame
- PCR:
-
Polymerase chain reaction
- RT-PCR:
-
Reverse transcription polymerase chain reaction
- TE:
-
Transposable element
References
Abe H, Urao T, Ito T, Seki M, Shinozaki K, Yamaguchi-Shinozaki K (2003) Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) function as transcriptional activators in abscisic acid signaling. Plant Cell 15:63–78
Anaya N, Roncero MI (1995) Skippy, a retrotransposon from the fungal plant pathogen Fusarium oxysporum. Mol Gen Genet 249:637–647
Baumann K, De Paolis A, Costantino P, Gualberti G (1999) The DNA binding site of the Dof protein NtBBF1 is essential for tissue-specific and auxin-regulated expression of the rolB oncogene in plants. Plant Cell 11:323–334
Beguiristain T, Grandbastien MA, Puigdomenech P, Casacuberta JM (2001) Three Tnt1 subfamilies show different stress-associated patterns of expression in tobacco consequences for retrotransposon control and evolution in plants. Plant Physiol 127:212–221
Boeke DJ, Corces VG (1989) Transcription and reverse transcription of retrotransposons. Annu Rev Microbiol 43:403–434
Boyle B, Brisson N (2001) Repression of the defense gene PR-10a by the single-stranded DNA binding protein SEBF. Plant Cell 13:2525–2537
Chang L, Zhang Z, Yang H, Li H, Dai H (2007) Detection of strawberry RNA and DNA viruses by RT-PCR using total nucleic acid as a template. J Phytopathol 155:431–436
Farman ML, Tosa Y, Nitta N, Leong SA (1996) MAGGY, a retrotransposon in the genome of the rice blast fungus Magnaporthe grisea. Mol Gen Genet 251:665–674
Flavell AJ, Pearce SR, Kumar A (1994) Plant transposable elements and the genome. Curr Opin Genet Dev 4:838–844
Friesen PD, Nissen MS (1990) Gene organization and transcription of TED, a lepidopteran retrotransposon integrated within the baculovirus genome. Mol Cell Biol 10:3067–3077
García-Martínez J, Martínez-Izquierdo JA (2003) Study on the evolution of the Grande retrotransposon in the Zea genus. Mol Biol Evol 20:831–841
Goda H, Sawa S, Asami T, Fujioka S, Shimada Y, Yoshida S (2004) Comprehensive comparison of auxin-regulated and brassinosteroid-regulated genes in Arabidopsis. Plant Physiol 134:1555–1573
Gojobori T, Moriyama EN, Kimura M (1990) Molecular clock of viral evolution, and the neutral theory. Proc Natl Acad Sci USA 87:10015–10018
Grandbastien MA (1998) Activation of plant retrotransposons under stress conditions. Trends Plant Sci 3:181–187
Grandbastien MA, Spielmann A, Caboche M (1989) Tnt1, a mobile retroviral-like transposable element of tobacco isolated by plant cell genetics. Nature 337:376–380
Hagen G, Guilfoyle T (2002) Auxin-responsive gene expression: genes, promoters and regulatory factors. Plant Mol Biol 49:373–385
Hansen LJ, Chalker DL, Sandmeyer SB (1988) Ty3, a yeast retrotransposon associated with tRNA genes, has homology to animal retroviruses. Mol Cell Biol 8:5245–5256
Hirochika H (1993) Activation of tobacco retrotransposons during tissue culture. EMBO J 12:2521–2528
Hirochika H, Otsuki H, Yoshikawa M, Otsuki Y, Sugimoto K, Takeda S (1996) Autonomous transposition of the tobacco retrotransposon Tto1 in rice. Plant Cell 8:725–734
Kato H, Sriprasertsak P, Seki H, Ichinose Y, Shiraishi T, Yamada T (1999) Functional analysis of retrotransposons in pea. Plant Cell Physiol 40:933–941
Kimura Y, Tosa Y, Shimada S, Sogo R, Kusaba M, Sunaga T, Betsuyaku S, Eto Y, Nakayashiki H, Mayama S (2001) OARE-1, a Ty1-copia retrotransposon in oat activated by abiotic and biotic stresses. Plant Cell Physiol 42:1345–1354
Konieczny A, Voytas DF, Cummings MP, Ausubel FM (1991) A superfamily of Arabidopsis thaliana retrotransposons. Genetics 127:801–809
Kumar A, Bennetzen JL (1999) Plant retrotransposons. Annu Rev Genet 33:479–532
Kumar S, Nei M, Dudley J, Tamura K (2008) MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform 9:299–306
Liubomirskaia NV, Kim AI, IuV Il’in (2003) Retrotransposon MDG4 and its role in genetic instability of a mutator strain of Drosophila melanogaster. Genetika 39:164–172
Lonnig WE, Saedler H (2002) Chromosome rearrangements and transposable elements. Annu Rev Genet 36:389–410
Ma Y (2008) Dissertation for doctorate: isolation and detection of LTR retrotransposon sequences from strawberry genome. Senyang Agricultural University, China
Ma Y, Sun H, Zhao G, Dai H, Gao X, Li H, Zhang Z (2008) Isolation and characterization of genomic retrotransposon sequences from octoploid strawberry (Fragaria × ananassa Duch.). Plant Cell Rep 27:499–507
MacRae AF (1998) A pentamer-repeat-comtaining DNA sequence in Texas bluebonnet (Lupinus texensis Hook.). Genome 41:553–559
Manninen I, Schulman AH (1993) BARE-1, a copia-like retroelement in barley (Hordeum vulgare L.). Plant Mol Biol 22:829–846
Marlor RL, Parkhurst SM, Corces VG (1986) The Drosophila melanogaster gypsy transposable elements encodes putative gene products homologous to retroviral proteins. Mol Cell Biol 6:1129–1134
Maruyama-Nakashita A, Nakamura Y, Watanabe-Takahashi A, Inoue E, Yamaya T, Takahashi H (2005) Identification of a novel cis-acting element conferring sulfur deficiency response in Arabidopsis roots. Plant J 42:305–314
Mount SM, Rubin GM (1985) Complete nucleotide sequence of the Drosophila transposable element copia: homology between copia and retroviral proteins. Mol Cell Biol 5:1630–1638
Nehra NS, Kartha KK, Stushnoff C (1991) Nuclear DNA content and isozyme variation in relation to morphogenic potential of Strawberry (Fragaria × ananassa) callus cultures. Can J Bot 69:239–244
Nemhauser JL, Mockler TC, Chory J (2004) Interdependency of brassinosteroid and auxin signaling in Arabidopsis. PLoS Biol 2:e258
Pearce SR, Harrison G, Li d, Heslop-Harrison JS, Kumar A, flavell AJ (1996) The Ty1-copia group retrotransposons in Vicia species: copy number, sequence heterogeneity and chromosomal localization. Mol Gen Genet 250:305–315
Perlman PS, Boeke JD (2004) Molecular biology. Ring around the retroelement. Science 303:182–184
Pontaroli AC, Rogers RL, Zhang Q, Shields ME, Davis TM, Folat KM, SanMiguel P, Bennetzen JL (2009) Gene content and distribution in the nuclear genome of Fragaria vesca. Plant Genome 2:93–101
Rico-Cabanas L, Martínez-Izquierdo JA (2007) CIRE1, a novel transcriptionally active Ty1-copia retratransposon from Citrus sinensis. Mol Genet Genomics 277:365–377
Saitou N, Nei M (1987) The Neighbor-joining method: a new method for reconstructing of phylogenetic trees. Mol Biol Evol 4:406–425
SanMiguel P, Tikhonov A, Jin YK, Motchoulskaia N, Zakharov D, Melake-Berhan A, Springer PS, Edwards KJ, Lee M, Avramova Z, Bennetzen JL (1996) Nested retrotransposons in the intergenic regions of maize genome. Science 274:765–768
Shim S, Lee SK, Han JK (2000) A novel family of retrotransposons in Xenopus with a developmentally regulated expression. Genesis 26:198–207
Simovic N, Wolyn D, Jelenkovic G (1992) Sequence analysis of 18S ribosomal RNA gene in Fragaria × ananassa Duch. cultivated octoploid strawberry. Plant Mol Biol 18:1217–1220
Sprinzl M, Steegborn C, Hübel F, Steinberg S (1996) Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res 24:68–72
Suoniemi A, Narvanto A, Schulman AH (1996) The BARE-1 retrotransposon is transcribed in barley from an LTR promoter active in transient assays. Plant Mol Biol 31:365–376
Tahara M, Aoki T, Suzuka S, Yamashita H, Tanaka M, matsunaga S, Kokumai S (2004) Isolation of an active element from a high-copy-number family of retrotransposons in the sweetpotato genome. Mol Gen Genomics 272:116–127
Takeda S, Sugimoto K, Otsuki H, Hirochika H (1998) Transcriptional activation of the tobacco retrotransposon Tto1 by wounding and methyl jasmonate. Plant Mol Biol 36:365–376
Takeda S, Sugimoto K, Otsuki H, Hirochika H (1999) A 13-bp cis-regulatory element in the LTR promoter of the tobacco retrotransposons Tto1 is involved in responsiveness to tissue culture, wounding, methyl jasmonate and fungal elicitors. Plant J 18:383–393
Tapia G, Verdugo I, Yañez M, Ahumada I, Theoduloz C, Cordero C, Poblete F, González E, Ruiz-Lara S (2005) Involvement of ethylene in stress-induced expression of the TLC1.1 retrotransposon from Lycopersicon chilense Dun. Plant Physiol 138:2075–2086
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The Clustal_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Vaucheret H, Marion-Poll A, Meyer C, Faure JD, Marin E, Caboche M (1992) Interest in and limits to the utilization of reporter genes for the analysis of transcriptional regulation of nitrate reductase. Mol Gen Genet 235:259–268
White SE, Habera LF, Wessler SR (1994) Retrotransposons in the flanking regions of normal plant genes: a role for copia-like elements in the evolution of gene structure and expression. Proc Natl Acad Sci USA 91:11792–11796
Yañez M, Verdugo I, Rodrígruz M, Prat S, Ruiz-Lara S (1998) Highly heterogeneous families of Ty1/copia retrotransposons in the Lycopersicum chilense genome. Gene 222:223–228
Acknowledgments
This work was supported by National Natural Science Foundation of China (Grant No. 30871689) and Program for New Century Excellent Talents in University (NCET-07-0565).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
He, P., Ma, Y., Zhao, G. et al. FaRE1: a transcriptionally active Ty1-copia retrotransposon in strawberry. J Plant Res 123, 707–714 (2010). https://doi.org/10.1007/s10265-009-0290-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10265-009-0290-0