
Abstract
Betula maximowicziana is an ecologically and economically important tree species in Japan. In order to examine the phylogeographical pattern of the species in detail, maternally inherited chloroplast (cp) DNA variations of 25 natural populations of Betula maximowicziana and a total of 12 populations of three related species were evaluated by PCR-RFLP analysis. Two main haplotypic groups of B. maximowicziana populations (northern and southern) were detected, with the main boundary passing through the Tohoku region in northeastern Japan; in addition there was high genetic differentiation among the 25 populations studied (GST = 0.950, \( G_{\text{ST}}^{\prime } = 0. 9 7 7 \)). The phylogeographical pattern exhibited by B. maximowicziana was much more similar to that of alpine plants than to that of beech and oak. Comparison of the patterns of genetic structure obtained from the cpDNA with previously and newly acquired data on bi-parentally inherited nuclear DNA indicates that the nuclear genome was transferred via pollen from the northern haplotypic group to the southern group more frequently than it moved in the opposite direction. Although common haplotypes were detected among B. maximowicziana and the two related species examined, these haplotypes were not shared sympatrically, suggesting very rare hybridization among the species currently occurring in their natural populations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Recent advances in genetic analysis techniques have greatly enhanced our ability to detect genetic structure in the nuclear genomes of plant species (Heuertz et al. 2004a; Tsuda and Ide 2005; Magri et al. 2006; Hiraoka and Tomaru 2009). However, their nuclear genomes are not only subject to recombination but are also biparentally inherited, and are thus affected by both pollen and seed flows from contributory lineages. This can complicate attempts to decipher a species’ phylogeographical pattern, evolutionary history and gene flow patterns. In contrast, the chloroplast (cp) genome in angiosperms is usually maternally inherited, so phylogeographical patterns revealed by cp DNA-based markers are attributable solely to seed flow among populations. Furthermore, since mutation rates in the cp genome are low, cp markers are not affected by recombination (Palmé et al. 2003a; Heuertz et al. 2004a). Therefore, since seed flow among populations is usually considered to be more restricted than pollen flow, cpDNA-based markers generally exhibit higher levels of population differentiation than nuclear markers (Petit et al. 1993; Palmé et al. 2003a), and thus their use facilitates the reconstruction of ancestral lineages (Palmé et al. 2003a). Moreover, since the dynamics of gene transfer via seed and via pollen are different (e.g., Liepelt et al. 2002), comparison of genetic structures detected by maternal cp and biparental nuclear marker analyses allows detailed assessment of the differences between wide-scale seed and pollen flow dynamics.
Betula maximowicziana (Monarch birch) belongs to the Betulaceae, and is found in the cool temperate zone of Japan, from the eastern part of Honshu Island to Hokkaido (Ohwi 1965). The species is considered to be endemic and restricted to Japan. B. maximowicziana is a diploid (2n = 28), monoecious, wind-pollinated species that produces wind-dispersed seeds. It is a pioneer tree species that grows rapidly in open sites, such as gaps, and is often found in even-aged stands (Osumi and Sakurai 1997). B. maximowicziana plays an important role in the stability and sustainability of forest ecosystems in the cool temperate zone of Japan, not only as a major pioneer tree species but also as a long-lived dominant species (Watanabe 1989; see also Tsuda and Ide 2005 for a discussion of its ecological importance). B. maximowicziana also yields high quality wood, so its use as a commercial crop from natural forests is being promoted, and its seeds or seedlings are currently being commercially distributed and/or translocated with little or no regard for their provenance. Therefore, mainly for conservation purposes, the species’ phylogeographical structure has been examined in several studies using nuclear DNA markers such as random amplified polymorphic DNA (RAPD; Tsuda et al. 2004) and simple sequence repeat (SSR) loci (Tsuda and Ide 2005). In the latter study, evidence from 23 populations was obtained indicating that they could be divided into two distinct groups, one located in the northern part of the species’ range and the other in the south. The two groups probably originated from populations that occupied different refugia during past glacial periods (Tsuda and Ide 2005). However, the groups identified were not clearly differentiated, partly because of the limitations of nuclear DNA for such analyses as a result of its bi-parental inheritance and extensive gene flow among populations, especially via pollen. In the current study, we examined maternal cpDNA variation in B. maximowicziana in the hope that it may reveal aspects of phylogeographical patterns that are difficult or impossible to detect using nuclear DNA markers. Our aim was to elucidate the phylogeography of the species at a higher resolution than has been achieved previously.
In addition, it has become increasingly clear that many plant species or species complexes share cpDNA haplotypes (e.g., Quercus, Petit et al. 2002; Okaura et al. 2007; Salix, Palmé et al. 2003b; Betula, Palme et al. 2004; Maliouchenko et al. 2007 and Fraxinus, Heuertz et al. 2006). In particular, Palme et al. (2004) and Maliouchenko et al. (2007) found evidence of extensive sharing of chloroplast haplotypes due to introgression in three European species of Betula: B. pendula, B. pubescens and B. nana. Since such extensive sharing of haplotypes among species disrupts species-specific phylogeographical patterns (Palme et al. 2004), we also need to consider the possible occurrence of haplotype sharing and introgression in B. maximowicziana and other Japanese birches in order to determine whether the phylogeographical pattern in B. maximowicziana detected by cpDNA is species-specific or influenced by extensive introgression among related species. For this reason, the cpDNA variations of three other birch species (B. ermani, B. platyphylla and B. grossa) were also assessed. According to the classification and taxonomy presented by de Jong (1993)—which has been used frequently in recent phylogenetic and taxonomic studies of Betula—B. maximowicziana, B. ermanii, B. platyphylla and B. grossa are members of the subgenera Betulaster, Neurobetula, Betula and Betulenta, respectively; thus, each of the four species examined here belongs to a different subgenus. Eleven Betula species occur naturally in Japan, but these three were selected for chloroplast DNA variation comparisons with B. maximowicziana because they are widely distributed, while the other species have much more restricted distributions. B. ermanii is a tetraploid (2n = 56) species found in the subalpine zone from Shikoku to Hokkaido; it usually inhabits sites at altitudes higher than those of other Betula species. B. platyphylla, like B. maximowicziana, is a diploid species (2n = 28) distributed from the eastern part of Honshu to Hokkaido. B. grossa is a hexaploid species (2n = 84) that is distributed from Kyushu to the northern part of Honshu Island and is not found in Hokkaido. Although each birch species has a unique life-history and a specific niche, these species often coexist in the same stands. A chemotaxonomic analysis by Keinänen et al. (1999) indicated that the four species examined here are relatively divergent from each other, but their degree of molecular genetic divergence and their apparent phylogeny depends on the sequence region and genome examined (Järvinen et al. 2004; Nagamitsu et al. 2006). Thus, the phylogenies derived from molecular data relating to nuclear and cpDNA in previous studies are not consistent (see e.g., Järvinen et al. 2004; Nagamitsu et al. 2006).
In this study, the cpDNA phylogeography of B. maximowicziana was assessed by PCR-RFLP analysis in an attempt to obtain clearer evidence of a major genetic dichotomy between the northern and southern groups. The species’ past colonization and gene flow patterns were also assessed using the data obtained from both cp and nuclear DNA analyses. As well as presenting the results of these analyses, the phylogeographical structures of B. maximowicziana and other tree species in Japan are compared herein, and related to their apparent persistence or disappearance in the northern region during the last glacial period.
Materials and methods
Plant sampling
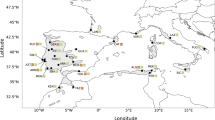
DNA samples from 16 randomly selected individuals from each of 25 natural populations covering most of the range of B. maximowicziana were used in the analysis (Fig. 1); 23 of these were sampled in our previous study (Tsuda and Ide 2005) and 2 (Erimo; H6 and Nanae; H7) were sampled in this study. Thus, samples from 400 individuals were used in total.

Locations and distributions of detected chloroplast haplotypes of the 25 Betula maximowicziana populations and 11 populations of three related species. Dashed boxes Locations at which representatives of more than two species were examined. For these populations, Be, Bp and Bg indicate the haplotypes of B. ermanii, B. platyphylla and B. grossa, respectively. No such abbreviations are used for B. maximowicziana. The code letters C, K, H and T followed by numerals refer to the B. maximowicziana populations examined in Chubu, Kanto, Hokkaido and Tohoku
To analyze the extent and patterns of haplotype sharing between B. maximowicziana and other Japanese birches, samples were also collected from eight individuals representing three to five populations of each of the species B. ermanii, B. grossa, and B. platyphylla. These three species were sampled from the same locations, or close to the locations, where B. maximowicziana was sampled.
DNA extraction and PCR-RFLP analysis
For the 2 newly sampled populations of B. maximowicziana and the 11 populations representing the other three species, total genomic DNA was extracted from fresh cambium or bud samples using a DNeasy Plant Mini Kit (Qiagen, Tokyo, Japan). For the other 23 populations, DNA extracted during our previous study (Tsuda and Ide 2005) was used. Three primer pairs—AS, CD and TF developed by Taberlet et al. (1991), Demesure et al. (1995) and Dumolin-Lapegue et al. (1997), respectively—and two restriction enzymes (HifI and TaqI) were used for the initial screening of the primer-enzyme combinations. This was conducted using 24 individuals sampled from geographically distant populations of B. maximowicziana and small samples from the other three species. Since TF exhibited low levels of variation and HifI and TaqI seemed to detect the same indel mutations in the TF region in this screening experiment, only the TF-TaqI combination was used for further analysis of this region.
The targeted sequences in each sample were amplified by PCR in 15 μL reaction mixtures containing 1–10 ng genomic DNA, 2.0 μM of each dinucleotide, 1× buffer, 2.5 mM MgCl2, 0.5 units LA Taq polymerase (Takara), and 0.2 μM of each primer in a DNA thermal cycler (Takara). The amplification program consisted of a denaturation step at 94°C for 5 min, followed by 45 cycles of 94°C for 30 s, annealing temperature for 30 s, 72°C for 4 min and finally 72°C for 5 min as a final extension step. The annealing temperatures (50, 55 and 54°C for the TF, CD and AS primers, respectively) and extension time were identical to those applied by Palmé and Vendramin (2002), but unlike the latter authors we did not use a touch-down PCR procedure. Each PCR product was cut with two restriction enzymes, HifI and TaqI (Promega, Madison, WI) in incubations at 37°C for HifI and 65°C for TaqI lasting 1.5–3 h in reaction mixtures with a total volume of 6.0 μL, containing 4.8 μL PCR product, 0.39 μL H2O, 1× buffer (Promega), 0.06 μL BSA (10 mg/mL) and 1.5 U of the respective enzyme. The resulting products were separated electrophoretically on 3.0–4.5% agarose gels in 1× TAE buffer. The gels were then stained with ethidium bromide, photographed on a UV transilluminator and the fragment variation was evaluated.
Analysis of PCR–RFLP variation
The gene diversity (H) of each population and the population differentiation (G ST) of each species were calculated. In addition, the population differentiation of B. ermanii was evaluated in terms of both unordered alleles (G ST) and ordered alleles (N ST), as described by Pons and Petit (1996), using the Permut software (developed by Petit, http://www.pierroton.inra.fr/genetics/labo/Software/Permut/index.html); 1,000 random haplotype permutations among populations were used to test whether the N ST values were significantly higher than the G ST values. According to Pons and Petit (1996), the presence of phylogeographical structure is indicated when the N ST values obtained are significantly higher than the G ST values. This test was performed only for B. ermanii because extremely little, or no, intra-population variation was detected in B. maximowicziana, B. grossa and B. platyphylla (see Results). Genetic variation between B. maximowicziana and B. ermanii was evaluated hierarchically by analysis of molecular variance (AMOVA: Excoffier et al. 1992) using GenAlEx ver. 6 software (Peakall and Smouse 2006: hereafter, GenAlEx). The significance of genetic variation, subdivided into between species, among populations within species, and among individuals within populations, was evaluated by a permutation test (n = 999) using GenAlEx. In addition, AMOVA was performed to evaluate the genetic variation among populations and among individuals within populations for the two species B. maximowicziana and B. ermanii. In this case, the significance of genetic variation was tested among populations using GenAlEx.
A haplotype network was constructed and edited following the statistical parsimony procedure implemented in TCS ver.1.06 software (Clement et al. 2000).
Comparison of within-haplotypic group genetic structures of B. maximowicziana using previous nuclear SSR data
The wide-scale genetic structure of B. maximowicziana was re-analyzed in detail using cpDNA data obtained in this study and information on variations at 11 nuclear SSR loci acquired from the two newly sampled and analyzed populations (32 individuals from the Nanae population and 45 individuals from the Erimo population) and the previous SSR dataset covering 1,014 individuals in 23 populations. The characteristics of these 11 loci, and the procedures used for the PCR genotyping, are described in Tsuda and Ide (2005). Using the genotype data for the 1,091 individuals in total, representing 25 populations, the average subpopulation gene diversity parameter H S and the population differentiation parameters G ST and F ST (Weir and Cockerham 1984) were calculated using FSTAT ver. 2.9.3.2 software (Goudet 2001: hereafter, FSTAT). The significance of F ST was tested by comparison to 95 and 99% confidence intervals derived from 1,000 bootstrap permutations implemented in FSTAT. The results of this study revealed two divergent cpDNA haplotypic population groups in B. maximowicziana (see Results). The genetic differentiation at the nuclear SSR loci associated with the two cpDNA haplotype population groups was evaluated hierarchically by AMOVA using Arlequin ver. 3.1 (Excoffier et al. 1992, 2007). To evaluate the genetic diversity within each cpDNA population group in more detail, the genetic diversity of nuclear SSR loci within each group was compared in terms of the parameters allelic richness (El Mousadik and Petit 1996), expected heterozygosity (H E) and F ST. The differences between groups, for individual parameters, were examined using a permutation test in FSTAT.
Standardization of population differentiation and pollen/seed migration ratios
Since absolute values of the population differentiation parameters F ST or G ST depend on the level of genetic variation in the examined material (Hedrick 2005), in this study, we also calculated standardized values of G ST, \( G_{\text{ST}}^{\prime } \) (Hedrick 2005), which always range from 0 to 1, using values of intra-population gene diversity (H S), total gene diversity (H T) and G ST. Thus, the measure allows comparisons between loci with different levels of genetic variation and allows us to examine genetic differentiation of organisms with different effective population sizes (Hedrick 2005).
The pollen/seed migration ratio, r (Ennos 1994), was calculated using the formula:
where m p is the pollen migration rate, m s the seed migration rate, G STc is the cytoplasmic G ST and G STn is the nuclear G ST. In this study, standardized \( G_{\text{STc}}^{\prime } \) and \( G_{\text{STn}}^{\prime } \) values were calculated for all materials used in the analysis, from either newly acquired data or data presented in the cited studies.
Results
Chloroplast DNA variation in B. maximowicziana and three related species
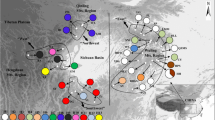
In total, 11 haplotypes were detected among the four species. In the analysis of the 25 B. maximowicziana populations, a total of four haplotypes were detected (Fig. 1; Table 1), and a total of nine haplotypes were detected in the analyses of the three related species: seven, two and one for B. ermanii, B. grossa and B. platyphylla, respectively (Fig. 1; Table 2). The identified haplotypes are described in Table 3 and the genetic relationships among the haplotypes, based on estimated mutational steps, are shown in Fig. 2. The B. maximowicziana populations in the northern (northern Tohoku and Hokkaido) and southern (southern Tohoku, Kanto and Chubu) areas were dominated by haplotypes A and D, respectively. Haplotype B was rare and unique to the Iwaizumi (T6) population. Haplotype C was extremely rare and detected in only one individual in the Naruko (T7) population. Thus, the group of populations dominated by haplotype A (H1 to T6 populations) is hereafter referred to as the “northern group” and the populations dominated by haplotype D (T7 to C6 populations) as the “southern group”. The Iwaizumi (T6) population is the only one that was found to include both the northern haplotype A and the southern haplotype D, although the frequency of haplotype D therein was low. Overall, for the 25 populations, the values of G ST and \( G_{\text{ST}}^{\prime } \) were 0.950 and 0.977, respectively, indicating that population differentiation was high.

Relationships between chloroplast haplotypes based on a statistical parsimony network. Circle sizes roughly reflect haplotype frequency, and thick lines in the network indicate mutational steps. The relationships between nine haplotypes (A–I) could be inferred (a). However, it was impossible to infer which of these haplotypes was most closely related to the remaining two haplotypes (J and K) in B. grossa (b), since no polymorphisms were shared between haplotypes A–I and J–K
For the related species examined, both intra-species and intra-population variations were detected only in B. ermanii (H T = 0.822, H S = 0.393). The N ST value for B. ermanii was 0.378 and was not significantly larger than its G ST value (=0.522). Further, although the G ST value for B. ermanii was not very high, the standardized parameter \( G_{\text{ST}}^{\prime } , \) taking variation within populations into consideration, was high (0.944), indicating that between-population differentiation was high in B. ermanii. The two haplotypes J and K were detected only in populations of B. grossa, and they diverged considerably from haplotypes (A–I) detected in B. maximowicziana, B. ermanii and B. platyphylla, differing in each case by at least eight mutational steps (Table 3). Although haplotype sharing among B. maximowicziana, B. ermanii and B. platyphylla was detected in this study (Fig. 1), the geographical distributions of the shared haplotypes differed among the species. In Otaki (K3), for example, B. ermanii population was dominated mostly by haplotype A, while the haplotype of all examined members of the B. maximowicziana population was haplotype D. In Towadako (T2), conversely, B. ermanii population was dominated mostly by haplotype D, while the haplotype of all examined members of the B. maximowicziana population was haplotype A.
The results of the hierarchical AMOVA of cpDNA data revealed that the proportions of genetic variance partitioned between the two species B. maximowicziana and B. ermanii, and among populations within these species were 14.55 and 76.63%, respectively, with the remainder (8.82%) occurring within-populations (Table 4). Each value of partitioned genetic variance was significant (P < 0.001) and, thus, these AMOVA results indicate significant genetic differentiation of the cpDNA among the two species. Although genetic variation among populations was significant for both of the species examined in the intra-species AMOVA, the amount of genetic variation subdivided into ‘among populations’ and ‘among individuals within populations’ was clearly different for B. maximowicziana and B. ermanii (Table 4).
Comparison of within-haplotypic group genetic structures of B. maximowicziana using previously acquired nuclear SSR data
The F ST value obtained from the analysis of nuclear SSRs in the samples from the 1,091 individuals representing 25 populations was 0.060, with confidence intervals of 0.049–0.073 (95%) and 0.047–0.077 (99%), indicating that population genetic differentiation was relatively low. Accordingly, based on the H S and G ST values in each nuclear SSR locus, the \( G_{\text{ST}}^{\prime } \) value was 0.100. The pollen/seed migration ratio r of B. maximowicziana was 387.5. The AMOVA of the nuclear SSR data revealed that the proportions of genetic variance partitioned between the two haplotypic groups of B. maximowicziana populations (northern and southern) and among populations within the groups were 2.32 and 4.69%, respectively, with the remainder (92.99%) occurring within-populations. Each value of partitioned genetic variance was significant (P < 0.001). None of the three parameters allelic richness, H E and F ST of nuclear SSR loci provided any indication of significant differences between the cpDNA population groups.
Discussion
Population differentiation of B. maximowicziana
The cpDNA variation detected here was clearly structured, with a single haplotype fixed in all of the populations except Iwaizumi (T6) and Naruko (T7). Therefore, population differentiation in the cpDNA of B. maximowicziana was high (\( G_{\text{ST}}^{\prime } = 0.977 \)) and higher than in its nuclear DNA (\( G_{\text{ST}}^{\prime } = 0.100 \)). This high \( G_{\text{ST}}^{\prime } \) value for B. maximowicziana cpDNA is similar to values reported for other Japanese broad-leaved tree species, such as Fagus crenata (G ST = 0.963, \( G_{\text{ST}}^{\prime } = 0.996; \) Tomaru et al. 1998). Okaura et al. (2007) also detected a high level of differentiation among populations (G ST = 0.853, \( G_{\text{ST}}^{\prime } = 0.974 \)) of Quercus mongolica var. crispula. On the other hand, according to estimates based on data presented by Palmé et al. (2003a), cpDNA population differentiation was lower in B. pendula (G ST = 0.42, \( G_{\text{ST}}^{\prime } = 0.696 \)) than in B. maximowicziana (which we investigated using the same primer–enzyme combinations as the cited authors; all the \( G_{\text{ST}}^{\prime } \) values were calculated by Y.T.).
Comparative phylogeography with B. maximowicziana
The main boundary associated with the cpDNA haplotypes of B. maximowicziana passes through the Tohoku region in northeastern Japan. Although identifying likely refugia and colonization patterns of individual Betula species in Japan using pollen fossil data would be difficult due to the difficulty of distinguishing between Betula species in pollen analysis, the phylogeographical patterns exhibited by B. maximowicziana and described here can be discussed on the basis of a comparative phylogeographical approach, comparing them with those of other species. On the one hand, although B. maximowicziana occasionally coexists with Fagus crenata and Q. mongolica var. crispula in cool temperate forests in Japan, phylogeographical patterns of these two species are different from that of B. maximowicziana. For example, F. crenata populations can be broadly divided into two groups: one on the Sea of Japan side and the other on the Pacific Ocean side of Japan, with a boundary running through Honshu Island. This pattern is apparent not only in the nuclear, cp and mitochondrial (mt) DNA variations (e.g., Tomaru et al. 1998; Fujii et al. 2002; Okaura and Harada 2002; Hiraoka and Tomaru 2009) but also in the morphological variations of the leaves (Hagiwara 1977). In Q. mongolica var. crispula, two main cpDNA lineages of the species have been identified, although their main boundary is further south than that of B. maximowicziana, running across central Honshu, south of the Tohoku region (Okaura et al. 2007). These differing phylogeographical patterns between B. maximowicziana and the other two tree species might mirror differences in the life-history traits of the species and different past colonization episodes influenced by species-specific life history traits, in particular cold-tolerance, as discussed by Lascoux et al. (2004). Since B. maximowicziana is cold tolerant, the species is considered to have survived in more northerly regions during past ice ages than other tree species such as beech and oak. Indeed, Bhagwat and Willis (2008) have presented evidence of various species’ ability to persist in northerly refugia based on the life-history traits of 12 European woody species of small-seeded, wind-dispersed angiosperms and gymnosperms—including Alnus, Betula, Picea, Pinus and Salix spp.
On the other hand, the phylogeographical pattern exhibited by B. maximowicziana is very similar to that of the alpine tree Pinus pumila, examined by Tani et al. (1996). P. pumila populations can be divided between the northern Tohoku and southern Tohoku regions largely on the basis of their allozymes (Tani et al. 1996). The similarity of these phylogeographical patterns between the cold-tolerant broadleaved tree, Betula pendula and the cold-tolerant conifer, Picea abies has also been discussed by Palmé et al. (2003a). Moreover, based on cpDNA lineages, similar genetic boundaries between the central Honshu and Tohoku regions have also been detected in five alpine plants: Pedicularis chamissonis, Primula cuneifolia, Loiseleuria procumbens, Cardamine nipponica, and Anemone narcissiflora (Fujii and Senni 2006). Fujii et al. (1997) and Fujii and Senni (2006) hypothesized that the most likely explanation for the phylogeographical pattern detected in alpine plants was that two major lineages had been transported southwards to the Japanese Archipelago during different glacial periods. The phylogeographical pattern exhibited by B. maximiwicziana might also be the result of multiple colonization events following different glacial episodes within the Japanese Archipelago, during repeated glacial and inter-glacial cycles. Alternatively, the two haplotypic groups detected here may represent different lineages originating from two distinct refugia established during a single glacial period. Although we cannot determine which of the two hypotheses better explains the observed phylogeographic pattern, both of these hypotheses suggest northerly persistence of B. maximowicziana during a glacial period, which is consistent with the species’ aforementioned cold-tolerance. In addition, private haplotypes were detected in the Iwaizumi (T6; haplotype B) and Naruko (T7; haplotype C) populations, suggesting that the areas around these populations may have been “cryptic refugia (Provan and Bennett 2008)” during a past ice age.
There were three mutations between the northern (A) and southern (D) haplotype of B. maximowiciziana in this study. Although the exact mutation rate of cpDNA in the Betulaceae is unknown, the fragment pattern of the PCR-RFLP examined suggests that these three are probably point mutations. The northern and southern haplotypes may have diverged over a time scale of millions of years, based on the fact that the substitution rate of rbcL in the cpDNA in the Fagaceae is 2.36 ± 0.79 × 10−10 (Frascaria et al. 1993), as also discussed in Okaura and Harada (2002) and Okaura et al. (2007). B. maximowicziana is endemic to Japan and a number of B. maximowicziana fossils from the Miocene period (Tertiary) have been found there (e.g., see the fossil lists in the homepages of the National Museum of Nature and Science, http://www.kahaku.go.jp/education/specimen_rent/kaseki.html, and the Gunma Museum of Natural History, http://www.gmnh.pref.gunma.jp/storageo/YB00001702/pageYB00001702.html). Therefore, although the phylogeography of tree species has been discussed mainly in the context of recolonization patterns following the last glacial maximum (LGM), detected cpDNA haplotypes and their distribution patterns in B. maximowicziana might have occurred and formed over a long time, and may have pre-dated the LGM. In support of this, recent studies in Europe suggest that longer temporal scales and episodes, such as repeated ice ages in the Quaternary or plate tectonic events in the Tertiary, should also be considered when discussing factors affecting modern phylogeographical structure (e.g., Magri et al. 2006, 2007; Ingvarsson 2008; Lascoux et al. 2008).
Gene transfer between northern and southern haplotypic groups
In previous nuclear SSRs analysis of B. maximowicziana (Tsuda and Ide 2005), two northern and southern clusters (assumed to be demes) were detected using Bayesian clustering STRUCTURE analysis developed by Pritchard et al. (2000). In the cited study, the populations in Hokkaido and the northern Tohoku region (H1–5 and T1–T6) belong to the northern cluster and the populations in the Kanto region and Central Honshu (K1–3 and C1–6) to the southern cluster. The STRUCTURE analysis indicated that both clusters accounted for ca. half of the ancestry of some populations located in the geographically intermediate zone (the southern Tohoku region between northern Tohoku and the northern central Honshu region) such as the Naruko (T7), Zao (T8) and Katsurao (T9) populations. These results show that geographically intermediate populations are also genetically intermediate according to the bi-parentally inherited nuclear DNA analysis. However, the maternal cpDNA variation of these populations was clearly structured, and their cpDNA haplotypes clearly belong to the southern group (with haplotype D predominating). These findings may reflect differences in gene flow dynamics between populations originating from northern and southern lineages, especially via pollen. In particular, these patterns may be influenced by past population colonization and admixing. For example, if the southern populations traveled rapidly northwards to the southern Tohoku region in lower densities than the northern population established there, the few southern immigrants could suffer from pollen limitation and might often receive pollen originating from northern populations. Since these areas are considered to represent the contact zone between northern and southern groups, such asymmetrical nuclear transfer and exchange via pollen will have occurred repeatedly and been complicated by hybridization between the groups and further backcrossing and/or hybridization between hybrids and purely southern or purely northern individuals. In contrast, clear separation between northern and southern cp genome in these areas suggests that transfer and exchange of the nuclear genome from populations in the northern Tohoku region (which have northern haplotypes) via seed to populations in the southern Tohoku region (which have southern haplotypes) has been less frequent than pollen flow.
Although the \( G_{\text{ST}}^{\prime } \)-based pollen/seed migration ratio, r, of B. maximowicziana (387.5) we obtained is lower than the reported value for Carpinus betulus (849.8, Grivet and Petit 2003; Coart et al. 2005), it appears to be substantially higher than that of several other tree species, for example B. pendula (287.2, Palmé et al. 2003a; Rusanen et al. 2003), Fagus sylvatica (60.6, Demesure et al. 1996; Comps et al. 2001), Fraxinus excelsior (54.1, Heuertz et al. 2004a, b) and Quercus suber (44.6, Toumi and Lumaret 1998; Belahbib et al. 2001) (all \( G_{\text{ST}}^{\prime } \)-based r values calculated by Y.T.). Thus, the potential pollen flow to potential seed flow ratio of B. maximowicziana seems to be within the higher part of the range for tree species. Therefore, the high value of the pollen/seed migration ratio of B. maximowicziana supports the hypothesis that pollen flow dominates in gene transfer events between the northern and southern groups.
Haplotype sharing among species
Ancestral polymorphism and introgression/hybridization are considered to be the main factors associated with the sharing of cpDNA haplotypes among related species. These two factors can be distinguished by estimating common ancestral haplotypes and their geographical distributions among species (Palme et al. 2004). Ancestral haplotypes are common among species (Watterson and Guess 1977); therefore, if the shared haplotypes are rare among species and are peripheral in the haplotype network, ancestral polymorphism is unlikely to provide an adequate explanation for the sharing (Palme et al. 2004). In addition, if the species shared haplotypes only as a result of ancestral polymorphism, their geographical distribution patterns should be independent of each other (Palme et al. 2004). Therefore, if hybridization and introgression had occurred frequently, both common and rare haplotypes should theoretically be shared, and show similar geographical patterns among species, as observed among B. pendula, B. pubescens and B. nana in central Europe by Palme et al. (2004). In contrast, common haplotypes, especially haplotype A were shared among the Japanese birches, and the geographical patterns of the shared haplotypes seem to be more species-specific than those of the European species, although limited numbers of populations were examined here. Therefore, AMOVA did show significant genetic variation among Japanese species in this study, whereas it did not detect significant genetic variation among species of European birches (Palme et al. 2004; Maliouchenko et al. 2007), suggesting that the species-component of genetic variation was stronger than the geographic component for Japanese birch species; European species exhibit the opposite pattern (Palme et al. 2004). These results suggest that current sympatric introgression/hybridization among species is very rare although ancestral polymorphism cannot be excluded as a possible factor explaining the cpDNA haplotype sharing detected in this study. Such rare hybridization is supported by our field observation that phenotypic hybrids between Japanese birches occur very rarely in natural populations (authors’ personal observations). However, further extensive sampling of Japanese birches, analysis of DNA sequences, physiological, palynological and palaeogeographical data are needed not only to elucidate the factors and processes that have shaped their haplotype sharing (e.g., ancestral polymorphism, ancient introgression and/or recent local hybridization–introgression) but also to draw clear conclusions about the population history of B. maximowicziana, including asymmetrical gene flow in the contact zone.
References
Belahbib N, Pemonge M-H, Ouassou A, Sbay H, Kremer A, Petit RJ (2001) Frequent cytoplasmic exchange between oak species that are not closely related: Quercus suber and Q. ilex in Morocco. Mol Ecol 10:2003–2012
Bhagwat SA, Willis KJ (2008) Species persistence in northerly glacial refugia of Europe: a matter of chance or biogeographical traits? J Biogeogr 35:464–482
Clement MD, Posada MD, Crandall KA (2000) TCS: A computer program to estimate gene genealogies. Mol Ecol 9:1657–1660
Coart E, Glabeke SV, Petit RJ, Bockstaele EV, Roldán-Ruiz I (2005) Range wide versus local patterns of genetic diversity in hornbeam (Carpinus betulus L.). Conserv Genet 6:259–273
Comps B, Gömöry D, Letouzey J, Thiébaut B, Petit RJ (2001) Diverging trends between heterozygosity and allelic richness during postglacial colonization in European beech. Genetics 157:389–397
De Jong PC (1993) An introduction to Betula: its morphology, evolution, classification and distribution, with a survey of recent work. In: Hunt D (ed) Proceedings of the IDS Betula symposium, 2–4 October 1992. International Dendrology Society, Richmond, UK
Demesure B, Sodzi N, Petit RJ (1995) A set of universal primers for amplification of polymorphic non-coding regions of mitochondrial and chloroplast DNA in plants. Mol Ecol 4:129–131
Demesure B, Comps B, Petit RJ (1996) Chloroplast DNA phylogeography of the common beech (Fagus sylvatica L.) in Europe. Evolution 50:2515–2520
Dumolin-Lapegue S, Pemonge M-H, Petit RJ (1997) An enlarged set of consensus primers for the study of organelle DNA in plants. Mol Ecol 6:393–397
El Mousadik A, Petit RJ (1996) Chloroplast DNA phylogeography of the argan tree of Morocco. Mol Ecol 5:547–555
Ennos RA (1994) Estimating the relative rates of pollen and seed migration among plant populations. Heredity 72:250–259
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distance among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 131:479–491
Excoffier L, Laval G, Schneider S (2007) ARLEQUIN version 3.1: an integrated software package for population genetics data analysis. http://cmpg.unibe.ch/ software/arlequin3
Frascaria N, Maggia L, Michaud M, Bousquet J (1993) The rbcL gene sequence from chestnut indicates a slow rate of evolution in Fagaceae. Genome 36:668–671
Fujii N, Senni K (2006) Phylogeography of Japanese alpine plants: biogeographic importance of alpine region of Central Honshu in Japan. Taxon 55:43–52
Fujii N, Ueda K, Watano Y, Shimizu T (1997) Intraspecific sequence variation of chloroplast DNA in Pedicularis chamissonis Steven (Scrophulariaceae) and geographic structuring of the Japanese “alpine” plants. J Plant Res 110:195–207
Fujii N, Tomaru N, Okuyama K, Koike T, Mikami T, Ueda K (2002) Chloroplast DNA phylogeography of Fagus crenata (Fagaceae) in Japan. Plant Syst Evol 232:21–33
Goudet J (2001) FSTAT; a program to estimate and test gene diversities and fixation indices version 2.9.3. http://www.unil.ch/izea/softwares/fstat.html
Grivet D, Petit RJ (2003) Chloroplast DNA phylogeography of the hornbeam in Europe: Evidence for a bottleneck at the outset of postglacial colonization. Conserv Genet 4:47–56
Hagiwara S (1977) Clines on leaf size of beech Fagus crenata (in Japanese). Species Biol Res 1:39–51
Hedrick PW (2005) A standardized genetic differentiation measure. Evolution 59:1633–1638
Heuertz M, Hausman J-F, Hardy OJ, Vendramin GG, Frascaria-Lacoste N, Vekemans X (2004a) Nuclear microsatellites reveal contrasting patterns of genetic structure between western and southeastern European populations of the common ash (Fraxinus excelsior L.). Evolution 58:976–988
Heuertz M, Fineschi S, Anzidei M, Pastorelli R, Salvini D, Paule L et al (2004b) Chloroplast DNA variation and postglacial recolonization of common ash (Fraxinus excelsior L.) in Europe. Mol Ecol 13:3437–3452
Heuertz M, Carnevale S, Fineschi S, Sebastiani F, Hausman JF, Paule L, Vendramin GG (2006) Chloroplast DNA phylogeography of European ashes, Fraxinus sp. (Oleaceae): roles of hybridization and life history traits. Mol Ecol 15:2131–2140
Hiraoka K, Tomaru N (2009) Genetic divergence in nuclear genomes between populations of Fagus crenata along the Japan Sea and Pacific sides of Japan. J Plant Res 122:269–282
Ingvarsson PK (2008) Multilocus patterns of nucleotide polymorphism and the demographic history of Populus tremula. Genetics 180:329–340
Järvinen P, Palmé A, Morales O, Lännenpää M, Keinänen M, Sopanen T, Lascoux M (2004) Phylogenetic relationships of Betula species (Betulaceae) based on nuclear ADH and chloroplast matK sequences. Am J Bot 91:1834–1845
Keinänen M, Julkunen-Tiitto R, Rousi M, Tahvanainen J (1999) Taxonomic implications of phenolic variation in leaves of birch (Betula L.) species. Biochem Syst Ecol 27:243–254
Lascoux M, Palmé AE, Cheddadi R, Latta RG (2004) Impact of ice ages on the genetic structure of tree and shrubs. Phil Trans R Soc B 359:197–207
Lascoux M, Pyhäjärvi T, Källman T, Savolainen O (2008) Past demography in forest trees: what can we learn from nuclear DNA sequences that we do not already know? Plant Ecol Diversity 1:209–215
Liepelt S, Bialozyt R, Ziegenhagen B (2002) Wind-dispersed pollen mediates postglacial gene flow among refugia. Proc Natl Acad Sci USA 99:14590–14594
Magri D, Vendramin GG, Comps B, Dupanloup I, Geburek T, Gömöry D, Latałowa M, Litt T, Paule L, Roure JM, Tantau I, van der Knaap WO, Petit RJ, de Beaulieu JL (2006) A new scenario for the Quaternary history of European beech populations: palaeobotanical evidence and genetic consequences. New Phytol 171:199–221
Magri D, Fineschi S, Bellarosa R, Buonamici A, Sebastiani F, Schirone B, Simeone MC, Vendramin GG (2007) The distribution of Quercus suber chloroplast haplotypes matches the palaeogeographical history of the western Mediterranean. Mol Ecol 16:5259–5266
Maliouchenko O, Palmé AE, Buonamici A, Vendramin GG, Lascoux M (2007) Comparative phylogeography and population structure of European Betula species, with particular focus on B. pendula and B. pubescens. J Biogeogr 34:1601–1610
Nagamitsu T, Kawahara T, Kanazashi A (2006) Endemic dwarf birch Betula apoinensis (Betulaceae) is a hybrid that originated from Betula ermanii and Betula ovalifolia. Plant Spec Biol 21:19–29
Ohwi J (1965) Flora of Japan (in Japanese). Shinbundo, Tokyo
Okaura T, Harada K (2002) Phylogeographical structure revealed by chloroplast DNA variation in Japanese beech (Fagus crenata Blume). Heredity 88:322–329
Okaura T, Quang NC, Ubukata M, Harada K (2007) Phylogegraphic structure and late Quaternary population history of the Japanese oak Quercus mongolica var. crispula and related species revealed by chloroplast DNA variation. Genes Genet Syst 82:465–477
Osumi K, Sakurai S (1997) Seedling emergence of Betula maximowicziana following human disturbance and the role of buried viable seeds. For Ecol Manag 93:235–243
Palmé AE, Vendramin GG (2002) Chloroplast DNA variation, postglacial recolonization and hybridization in hazel, Corylus avellena. Mol Ecol 11:1769–1779
Palme AE, Su Q, Palsson S, Lascoux M (2004) Extensive sharing of chloroplast haplotypes among European birches indicated hybridization among Betula pendula, B. pubescens and B. nana. Mol Ecol 13:167–178
Palmé AE, Su Q, Rautenberg A, Manni F, Lascoux M (2003a) Postglacial recolonization and cpDNA variation of silver birch, Betula pendula. Mol Ecol 12:201–212
Palmé AE, Semerikov V, Lascoux M (2003b) Absence of geographic structure of chloroplast DNA variation in sallow, Salix caprea L. Heredity 91:465–474
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Petit RJ, Kremer A, Wagner DB (1993) Finite island model for organelle and nuclear genes in plants. Heredity 71:630–641
Petit RJ, Csaikl UM, Bordács S, Burg K, Coart E, Cottrell J et al (2002) Chloroplast DNA variation in European white oaks phylogeography and patterns of diversity based on data from over 2600 populations. For Ecol Manag 156:5–26
Pons O, Petit RJ (1996) Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 144:1237–1245
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Provan J, Bennett KD (2008) Phylogeographic insight into cryptic glacial refugia. Trends Ecol Evol 23:564–571
Rusanen M, Vakkari P, Blom A (2003) Genetic structure of Acer platanoides and Betula pendula in northern Europe. Can J For Res 33:1110–1115
Taberlet P, Gielly L, Pautou G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol 17:1105–1109
Tani N, Tomaru N, Araki M, Ohba K (1996) Genetic diversity and differentiation in populations of Japanese stone pine (Pinus pumila) in Japan. Can J For Res 26:1454–1462
Tomaru N, Takahashi M, Tsumura Y, Takahashi M, Ohba K (1998) Intraspecific variation and phylogeographic patterns of Fagus crenata (Fagaceae) mitochondrial DNA. Am J Bot 85:629–636
Toumi L, Lumaret R (1998) Allozyme variation in cork oak (Quercus suber L.): the role of phylogeography and genetic introgression by other Mediterranean oak species and human activities. Theor Appl Genet 97:647–656
Tsuda Y, Ide Y (2005) Wide-range analysis of genetic structure of Betula maximowicziana, a long-lived pioneer tree species and noble hardwood in the cool temperate zone of Japan. Mol Ecol 14:3929–3941
Tsuda Y, Goto S, Ide Y (2004) RAPD analysis of genetic variation within and among four natural populations of Betula maximowicziana. Silvae Genet 53:234–239
Watanabe S (1989) Silvical characteristics of Betula maximowicziana (in Japanese). For Tree Breed Hokkaido 32:15–18
Watterson GA, Guess HA (1977) Is the most frequent allele the oldest? Theor Pop Biol 11:141–160
Weir BS, Cockerham CC (1984) Estimating f-statistics for the analysis of population structure. Evolution 38:1358–1370
Acknowledgments
We are grateful to Jin Yamasaki, Makoto Kawano, Susumu Goto and Yasuo Takahashi for their help in sampling the plant material. We also thank the staff members of the University Forests, the University of Tokyo, for collecting samples from the Furano population in the University Forest in Hokkaido, and the Otaki population in the University Forest in Chichibu. We thank Professor Ko Harada for providing us with the manuscript by Okaura et al. (2007), which was then in press. This study was partly supported by Grants-in-Aid for Scientific Research (No. 19380082) from the Ministry of Education, Culture, Sports, Science and Technology, Japan and a grant for Research on Genetic Guidelines for Restoration Programs using Genetic Diversity Information from the Ministry of Environment, Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tsuda, Y., Ide, Y. Chloroplast DNA phylogeography of Betula maximowicziana, a long-lived pioneer tree species and noble hardwood in Japan. J Plant Res 123, 343–353 (2010). https://doi.org/10.1007/s10265-009-0280-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10265-009-0280-2