
Abstract
Introgression has been considered to be one of main factors leading to phylogenetic incongruence among different datasets at lower taxonomic levels. In the plants of Pinaceae, the mtDNA, cpDNA, and nuclear DNA (nrDNA) may have different evolutionary histories through introgression because they are inherited maternally, paternally and biparentally, respectively. We compared mtDNA, cpDNA, and two low-copy nrDNA phylogenetic trees in the genus Pinus subgenus Strobus, in order to detect unknown past introgression events in this group. nrDNA trees were mostly congruent with the cpDNA tree, and supported the recent sectional and subsectional classification system. In contrast, mtDNA trees split the members of sect. Quinquefoliae into two groups that were not observed in the other gene trees. The factors constituting incongruence may be divided into the following two categories: the different splits within subsect. Strobus, and the non-monophyly of subsect. Gerardianae. The former was hypothesized to have been caused by the past introgression of cpDNA, mtDNA or both between Eurasian and North American species through Beringia. The latter was likely caused by the chimeric structure of the mtDNA sequence of P. bungeana, which might have originated through past hybridization, or through a horizontal transfer event and subsequent recombination.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
With the proliferation of molecular tools and accumulation of DNA sequence data, phylogenetic estimation using multiple datasets from independent genetic loci has become commonplace over the last decade. The congruence of multiple gene trees is the most important means of assessing the reliability of a hypothesis regarding organismal evolutionary relationships. Even if phylogenetic incongruence is observed, it may provide a window into valuable evolutionary processes that cannot be accessed with only one dataset. For example, the widespread occurrence of “cytoplasmic capture”, introgression of cytoplasmic genomes across species without concomitant introgression of nuclear genes, was first suggested by the unexpected contradictory results observed between chloroplast DNA (cpDNA) and morphology, and also between cpDNA and nuclear markers in plants (Rieseberg and Soltis 1991). Theoretically, incongruence between gene tree and species phylogeny, or between different gene trees, can result from a variety of evolutionary processes, such as orthology and paralogy conflation, lineage sorting of ancestral polymorphism, and introgressive hybridization (Wendel and Doyle 1998). Among these causes, introgressive hybridization has been found to be a prominent factor that leads to phylogenetic incongruence among different datasets at lower taxonomic levels (Cronn and Wendel 2004).
Pinus L. (Pinaceae)—the largest genus of conifers, comprising approximately 110 species—is distributed widely in the northern hemisphere. Due to the high economic utility of pine species, many artificial hybridization experiments have been conducted for breeding purposes. These trials revealed that hybridization could be successful in many interspecific combinations (Critchfield 1986; Garrett 1979). In addition to the artificial crossing data, several rigid instances of introgressive hybridization between sympatric pine species have been clarified by molecular analyses: P. banksiana and P. contorta in Canada and the northern United States (Dong and Wagner 1993; Wagner et al. 1987), P. taeda and P. echinata in the south-eastern United States (Chen et al. 2004), P. hartwegii and P. montezumae in Mexico (Matos and Schaal 2000), and P. pumila and P. parviflora in Japan (Watano et al. 2004). In the latter case of Pinus hybridization, one of the authors found that cpDNA introgression occurred uni-directionally from P. parviflora to P. pumila, while mitochondrial DNA (mtDNA) introgression occurred in the opposite direction, from P. pumila to P. parviflora (Watano et al. 1995, 1996, 2004). mtDNA introgression from P. pumila to P. parviflora is regionally extensive, and nearly all plants of P. parviflora collected in the southern part of Tohoku district, Japan, were found to have the mtDNA of P. pumila (Senjo et al. 1999; Tani et al. 2003). Independent patterns of introgression of two cytoplasmic genomes are possible in Pinus because their cpDNA and mtDNA are transmitted paternally and maternally, respectively (Neale and Sederoff 1989). The finding of the contrasting introgression pattern of cpDNA and mtDNA, and the example of regional fixation of heterospecific cytoplasmic genomes in hybridization between P. pumila and P. parviflora, have suggested that phylogenetic comparison of independently inherited genetic markers would be useful to detect cryptic introgression events among Pinus species.
The present study focuses on the phylogenetic relationships of the species in Pinus subgenus Strobus, including P. pumila and P. parviflora. Critchfield (1986) compiled the results of controlled interspecific crosses in white pines (subgenus Strobus section Strobus sensu Little and Critchfield 1969), and reported that one-half of the verified hybrids were crosses of Eastern and Western hemisphere species. These results are in contrast with the results of crosses in hard pines (subgenus Pinus section Pinus sensu Little and Critchfield 1969), in which the ability to hybridize is tied closely to geography. In this respect, subgenus Strobus was expected to be more suitable for our purpose than subgenus Pinus, because the cross-ability, even in geographically isolated species combinations, satisfies a prerequisite for unknown past episodes of introgressive hybridization through historical changes in geographical distribution.
The results of phylogenetic studies on subgenus Strobus using cpDNA (Eckert and Hall 2006; Gernandt et al. 2005; Wang et al. 1999) and the nuclear internal transcribed spacer (ITS) region of rDNA (Gernandt et al. 2001; Liston et al. 1999) sequences are broadly congruent. Based mainly on cpDNA phylogeny, Gernandt et al. (2005) revised the classification system of genus Pinus, and divided the members of subgenus Strobus into section Quinquefoliae, composed of three subsections (Strobus, Krempfianae, and Gerardianae), and section Parrya, composed of three subsections (Cembroides, Nelsoniae, and Balfourianae). Recently, Syring et al. (2007) examined the phylogenetic relationship among subgenus Strobus by using a nuclear late embryogenesis abundant (LEA)-like gene, with two or more alleles from each species. Monophyly of subsections defined by Gernandt et al. (2005) was also supported by this nuclear single-copy gene. However, the phylogenetic relationships within each subsection remain controversial. In sect. Quinquefoliae subsection Strobus, for example, all Eurasian members of the subsection except for P. peuce and one North American species, P. albicaulis, form a monophyletic group in the cpDNA tree, but not in the nuclear LEA-like gene tree. The incongruent species relationships between cpDNA and the LEA-like gene could be caused primarily by incomplete lineage sorting of the LEA-like gene, as suggested by Syring et al. (2007). However, the possibility of introgression of cpDNA and (or) the LEA-like gene cannot be excluded at this stage. Therefore, in order to detect the footprint of past introgression events, it is essential to test additional independent DNA markers such as other nuclear genes and maternal mtDNA. mtDNA sequence data of Pinus have been utilized mainly for phylogeographical studies (Burban and Petit 2003; Richardson et al. 2002) and the comparison of closely related species (Gugerli et al. 2001).
We present herein phylogenetic analyses of subgenus Strobus based on the usage of all three differently inherited genomic regions—maternal mtDNA, paternal cpDNA, and two nuclear single or low-copy genes—in order to draw inferences regarding past hybridization events. The results show that the recent sectional and subsectional classification system of subgenus Strobus (Gernandt et al. 2005) based on the cpDNA tree is supported by two nuclear gene trees, but not by the mtDNA tree. We analyze the differences in phylogenetic information among datasets to specify the taxa causing the incongruence, and discuss which of the evolutionary processes appears to be the cause of the incongruence.
Materials and methods
Taxon sampling and DNA extraction
We used 17 ingroup taxa, including 13 species from section Quinquefoliae subsection Strobus, two from sect. Quinquefoliae subsect. Gerardianae, one from sect. Parrya subsect. Cembroides, and one from sect. Parrya subsect. Balfourianae (Table 1). Our sampling focused mainly on section Quinquefoliae, which includes P. pumila and P. parviflora, and covered 60% of the species in this section, but lacked species from the monotypic subsection Krempfianae. Pinus canariensis and P. sylvestris from subgenus Pinus were used as outgroup taxa for cpDNA and nuclear DNA trees. In the analysis of mtDNA, P. ponderosa was used as one of the outgroup taxa instead of P. canariensis. Both nuclear DNA and cpDNA sequence data (Syring et al. 2005; Gernandt et al. 2005) showed that two subgenera of genus Pinus were monophyletic and sister to each other. One individual per each of the taxa has been examined. A sample of 100 mg fresh needles per individual was cut into small pieces of 2 mm in length and desiccated in silica-gel powder. Total DNA was extracted from these desiccated samples following the methods described in Suyama et al. (2000).
PCR amplification, sequencing and alignment
As for the mtDNA, we used two regions: a partial sequences of NADH dehydrogenase subunit 1 gene (nad1) intron 2 and NADH dehydrogenase subunit 5 gene (nad5) intron 1. Primers designed by Demesure et al. (1995) were used for the amplification of nad1 intron 2, and those by Wang et al. (2000) for nad5 intron 1. The thermal profiles for PCR of both regions were as follows: initial denaturation at 95°C for 3 min, 3 cycles at 94°C for 1 min, 62°C for 1 min, 72°C for 2 min, followed by 3 cycles at 94°C for 1 min, 60°C for 1 min, 72°C for 2 min, followed by 34 cycles at 94°C for 1 min, 58°C for 1 min, 72°C for 2 min, and a final extension at 72°C for 10 min. As for the cpDNA, four regions (rbcL, matK, trnV intron, rpl20-rps18) used by Wang et al. (1999) were amplified. PCR primers and the thermal profile for amplification were the same as those of Wang et al. (1999). As for nuclear DNAs, we chose the Pinus taeda expressed sequence tag (EST) locus PtIFG9008 and the plastid type NAD+ dependent glyceraldehydes-3-phosphate dehydrogenase gene (GapCp). PtIFG9008 was derived from Pinus taeda cDNA clone (INSD accession AA739854) for map construction (Temesgen et al. 2001). The cDNA sequence shows high similarity to Arabidopsis thaliana L. AAA-type ATPase family protein (AT4G02480). This region was amplified by primers from Temesgen et al. (2001). Primers for GapCp were newly designed based on the sequence of Pinus sylvestris (INSD accession AJ001706). The forward and reverse primers (5′-GCTTTCCGTGTACCAACACCCA-3′, 5′-CCCCACTCATTGTCATACCA-3′) are located in exons 9 and 11 of P. sylvestris GapCp, respectively. The thermal profiles for PCR of both genes were as follows: initial denaturation at 95°C for 3 min, 3 cycles at 94°C for 1 min, 62°C for 1 min, 72°C for 2 min, followed by 3 cycles at 94°C for 1 min, 59°C for 1 min, 72°C for 2 min, followed by 34 cycles at 94°C for 45 s, 56°C for 45 s, 72°C for 1 min 30 s, and a final extension at 72°C for 10 min. Because our DNA samples were from diploid sporophytic tissues, PCR products from heterozygous plants could not be used as templates for direct sequencing. In order to distinguish heterozygotes and homozygotes, we performed single-strand conformation polymorphism (SSCP) analysis following the method of Watano et al. (2004). PCR samples showing two-banded patterns in SSCP gels, which are typical for a homozygote, were subjected to direct sequencing. When samples showed three- or four-banded patterns in SSCP gels, which are indicative of a heterozygous plant, each band was cut out and the DNA of the band was extracted by using an E.Z.N.A. Poly Gel DNA Extraction kit (Omega Bio-tek, Doraville, GA). The extracted single-stranded DNA was amplified using the same PCR primers as those used to amplify from genomic DNA. The resulting PCR product was used as a sequencing template.
PCR products were purified using GeneClean II (Qbiogene, Irvine, CA) or ExoSAP-IT (GE Healthcare, Buckinghamshire, UK). Sequencing was carried out on an ABI 310 automated DNA sequencer using the BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems, Foster City, CA). Sequences were edited and assembled by AutoAssembler v.2.1 (Applied Biosystems) to construct a consensus sequence for each taxon. Multiple alignment was conducted by using ClustalX (Thompson et al. 1997) and subsequently adjusted manually in BioEdit (Hall 1999).
Phylogenetic and statistical analyses
All of the sequences used in each dataset and their International Nucleotide Sequence Database (INSD) accession number are listed in Table S1 (see Electronic Supplementary Materials). Phylogenetic analysis was performed by the maximum parsimony (MP) method with PAUP* version 4.0b10 (Swofford 2003), and the Bayesian method with MrBayes ver.3.1.2 (Ronquist and Huelsenbeck 2003). As with the MP analysis, all nucleotide sites with gaps were initially excluded, and insertions and deletions (indels) were not recorded. In the case of mtDNA, however, indels were coded using the simple indel coding method (Simmons and Ochoterena 2000) implemented in SeqState v.1.32 (Müller 2005). This is because the mtDNA intron sequences showed low variation at nucleotide substitutions and high levels of length variation. A heuristic search was performed with a zero branch-length collapse option, 100 random sequence addition, tree bisection–reconnection (TBR) branch swapping, and the “keeping multiple trees” (MulTrees) option in effect. The bootstrap analysis was conducted on 1,000 replicates, TBR branch swapping, and simple sequence addition. In the Bayesian analysis, we selected the best-fit model of nucleotide substitution for each dataset using PAUP* version 4.0b10 and MrModeltest 2.3 (Nylander 2004). Bayesian inference search was performed during 1 million generations with sampling trees every 100 generations using MrBayes ver.3.1.2. A majority consensus tree and posterior probability (PP) of each branch were obtained from the sampled trees with burn-in value setting at 2,500.
Congruence among sequence datasets was examined with the incongruence length difference (ILD) test (Farris et al. 1994), implemented in PAUP* v.4.0. In order to prepare counterpart sequences, Pinus ponderosa was deleted from the mtDNA dataset, and P. canariensis from the cpDNA and nrDNA datasets. Pinus kwangtungensis in the cpDNA dataset was used as a counterpart of P. fenzeliana in the mtDNA and nrDNA datasets. Nuclear DNA datasets contained two allelic sequences in some taxa when the samples were heterozygous. For the ILD test using the nrDNA datasets, we adjusted the numbers of sequences in both datasets by using the same sequence twice in another dataset (1+1 and 2+1). The ILD tests were performed with 1,000 replications of heuristic search with TBR branch swapping, and simple sequence addition. In order to evaluate the incongruence between gene trees, we additionally examined whether the number of steps required of a dataset under the constraints of well-supported nodes observed in the other dataset was significantly greater or not, by using the Templeton’s test (Templeton 1983) implemented in PAUP* v.4.0.
Results
mtDNA sequences and phylogeny
Partial nucleotide sequences of mitochondrial nad1 intron 2 were newly obtained from 14 taxa. The sequences of five taxa (P. cembra, P. pumila, P. sibirica, P. sylvestris and P. ponderosa) were from INSD. The second intron of nad1 is a group II intron, which is characterized by a uniform structure of six major domains radiating from a central wheel (Michel et al. 1989). The region sequenced was found to correspond to domains III and IV based on comparison with the alignment of stem regions (see Fig. 6 in Won and Renner 2003). The sequenced region showed high levels of length variation in Pinus, from 1,458 bp in P. koraiensis to 2,095 bp in P. aristata. Because of the extensive indels in domain IV, the aligned sequences were 4,219 bp in length, which is over twice the original sequence length. All of the nad5 intron 1 nucleotide sequences were determined for the first time in this study. The lengths of the nad5 intron 1 differed greatly between the two subgenera, but were relatively uniform within subgenera: from 1,221 to 1,226 bp in subgenus Strobus, and from 1,407 to 1,415 bp in subgenus Pinus. The ILD test showed that the incongruity between nad1 and nad5 was just not significant at the 5% level (P = 0.052). Thus, to avoid combining different phylogenetic information, both separate and combined datasets were analyzed. The aligned sequences of the combined dataset were 5,642 bp in length. The FASTA format file of the mtDNA combined dataset is given in the Electronic Supplementary Material (Table S2). The combined dataset with and without indel information contained 94 and 46 parsimony-informative (PI) characters, respectively.
MP analysis of the combined dataset without indels generated two equally parsimonious trees requiring 73 steps (CI = 0.82, RI = 0.92). The combined dataset with indels yielded 19,536 equally parsimonious trees requiring 189 steps (CI = 0.91, RI = 0.93). The strict consensus trees are shown in Figs. 1a and b. In both trees, 17 species of subgenus Strobus were split into three clades. The first clade (GROUP_1) consisted of six species of sect. Quinquefoliae subsect. Strobus (QS) and P. gerardiana of sect. Quinquefoliae subsect. Gerardianae (QG). The second clade (GROUP_2) was composed of seven species of subsect. Strobus (QS) and P. bungeana of subsect. Gerardianae (QG). Pinus bungeana was sister to the other members of this clade. The last clade (GROUP_3) included species of sect. Parrya. The separation of the two species from subsect. Gerardianae into different clades was not observed in any previous phylogenetic analyses of genus Pinus. Pinus strobus, P. pumila and P. koraiensis formed a relatively well-supported clade (88%) in the dataset with indels, but not in that without indels. Synapomorphies of this clade were four indel characters.

Strict consensus tree of the maximum parsimonious trees based on mitochondrial (mt)DNA sequences from 17 species of Pinus subgenus Strobus and two outgroups. Support values from the maximum parsimony (MP) and Bayesian methods are mapped on each branch (MP bootstrap/Bayesian posterior probability). Support values in Fig. 1b are only MP bootstrap probability. Abbreviations in parenthesis: QS section Quinquefoliae subsection Strobus, QG sect. Quinquefoliae subsect. Gerardianae, PC sect. Parrya subsect. Cembroides, PB sect. Parrya subsect. Balfourianae
The dataset of nad1 intron 2 included 34 PI sites, and the MP analysis generated seven equally parsimonious trees of 45 steps (CI = 0.87, RI = 0.96). Although the three clades in the combined dataset were resolved in the five of the seven shortest trees, the member of GROUP_1 was not grouped in the strict consensus tree (Fig. 1c). The dataset of nad5 intron 1 contained 12 PI sites, and we obtained two equally parsimonious trees of 25 steps (CI = 0.80, RI = 0.89) by MP analysis. As for the nad5 intron 1 dataset, the strict consensus tree was different from the nad1 tree and also from the combined dataset trees, in that P. bungeana was not included in GROUP_2 (Fig. 1d).
The Bayesian method for nad1, nad5 and the combined datasets without indels generated trees that were mostly congruent with those of the MP method (data not shown). In the Bayesian tree for the combined dataset without indels, the species of sect. Quinquefoliae (GROUP_1 and _2 in Figs. 1a, b) form a monophyletic clade with a posterior probability (PP) of 0.90.
cpDNA sequences and phylogeny
Sequences of the four cpDNA regions (rbcL, matK, trnV intron, rpl20-rps18) were newly determined from five species, P. albicaulis, P. cembroides, P. flexilis, P. sibirica, and P. strobiformis. The cpDNA sequences of P. fenzeliana were not determined. Instead, we used the sequences of P. kwangtungensis determined by Wang et al. (1999), which is sometimes treated as a synonym of P. fenzeliana (Price et al. 1998). The sequences of other taxa were obtained from INSD. The aligned sequences of the four cpDNA regions consisted of 3,489 sites, and contained 97 PI sites. The ILD test revealed that the four regions yielded trees not statistically different (P = 0.329). The MP analysis of the combined dataset generated two most parsimonious trees (tree length = 164, CI = 0.88, RI = 0.91). The phylogenetic relationship shown in the strict consensus tree (Fig. 2) was mostly congruent with the results of previous cpDNA studies in Pinus (Eckert and Hall 2006; Gernandt et al. 2005; Wang et al. 1999). The Bayesian tree was also identical to the MP consensus tree when the branches with PPs less than 0.60 were collapsed. The 17 species of subgenus Strobus were split into two groups corresponding to the sections Parrya (PB and PC) and Quinquefoliae (QS and QG). Within sect. Quinquefoliae, two species of subsect. Gerardianae (QG) formed a clade, which was sister to monophyletic subsect. Strobus (QS).

Strict consensus tree of the MP trees based on chloroplast (cp)DNA sequences from 17 species of Pinus subgenus Strobus and two outgroups. Support values from the MP and Bayesian methods are mapped on each branch (MP bootstrap/Bayesian posterior probability). The geographical distribution of each taxon and the clade defined in the text (Eurasian Clade) are shown by right brackets
Nuclear DNA sequences and phylogeny
The nucleotide sequences determined from PCR products of PtIFG9008 were from 475 to 478 bp in length and were easily aligned. The SSCP of PCR products from three samples, P. pumila, P. fenzeliana, and P. sibirica, showed heterozygous band-patterns, and so two allelic sequences were determined for these three species. As a result, the dataset of PtIFG9008 contained 22 sequences. The alignment with the P. taeda cDNA sequence (AA739854) of this gene suggested that the sequences of the PCR products included two introns. Exon regions of the alignment were 246 bp in total, and contained nine PI sites, while intron regions (234 bp in an alignment) contained 24 PI sites. The MP analysis generated six equally parsimonious trees requiring 52 steps (CI = 0.94, RI = 0.95). The strict consensus tree (Fig. 3a) was mostly congruent with the cpDNA tree (Fig. 2) except for the species relationship within subsect. Strobus. The Bayesian tree was mostly congruent with the strict consensus tree of the MP method. For the species in which two alleles were sequenced, P. pumila and P. fenzeliana exhibited allelic monophyly. The alleles of P. sibirica formed a group with an allele of P. cembra.

Strict consensus tree of the MP trees based on nuclear DNA sequences from 17 species of Pinus subgenus Strobus and two outgroups: a PtIFG9008 tree, b GapCp tree. Support values from the MP and Bayesian methods are mapped on each branch (MP bootstrap/Bayesian posterior probability). Different allelic sequences from an individual are designated as species epithet plus _1 or _2
We obtained an aligned sequence of 501 bp for GapCp, corresponding to positions 7,204–7,686 of P. sylvestris genomic sequences of GapCp1 gene (AJ001706). The sequences determined were the region from the 3′ part of exon 9 to the 5′ part of exon 11. Two introns were situated at positions 48–301 and 389–473 in an alignment, respectively. Two alleles were sequenced from five species, and thus the dataset contained 24 sequences from 19 species. Exons (162 bp) and introns contained 18 and 15 PI sites, respectively. The MP analysis generated only one parsimonious tree (Fig. 3b) requiring 75 steps (CI = 0.89, RI = 0.91). The Bayesian method also generated a tree identical to the strict consensus tree of the MP method. The tree supported the recent sectional and subsectional classification system based on the cpDNA tree. However, the phylogenetic relationship within subsect. Strobus was not well-resolved because of the non-monophyly of conspecific alleles.
ILD and Templeton’s tests among mtDNA, cpDNA, and nrDNA trees
We examined whether the four datasets contained different phylogenetic information using ILD tests (Table 2). Among six pairs, only cpDNA vs PtIFG9008 was congruent, while all other pairs were significantly incongruent. Topological differences between the mtDNA tree of the combined dataset (Figs. 1a, b) and the others (Figs. 2, 3) are apparent because P. gerardiana and P. bungeana of sect. Quinquefoliae subsect. Gerardianae were split into two different groups in the mtDNA tree. To confirm the causes of the incongruence, three topology constraints were adopted for each dataset. The first constraint is the split of GROUP_1 and GROUP_2 found in the combined datasets of mtDNA sequences (Figs. 1a, b). The second is a monophyly of the seven taxa of subsect. Strobus in GROUP_2, which was a phylogenetic signal held in the combined mtDNA and nad5 datasets (Figs. 1a, b, d). The cpDNA tree (Fig. 2) split the species of subsect. Strobus into two groups: P. albicaulis of North America together with the Eurasian species except P. peuce, and the other species. Hereafter, we will refer to the former monophyletic cpDNA group as the “Eurasian Clade.” This was the last constraint adopted. The results of the Templeton’s test are shown in Table 3. The first constraint led to significantly less parsimonious trees in the cpDNA and nrDNA datasets. The second and final constraints resulted in significantly less parsimonious trees in the cpDNA and mtDNA datasets, respectively, but not in the nrDNA datasets.
Discussion
Incongruence between mtDNA and other trees
CpDNA, PtIFG9008 and GapCp trees were congruent in the following respects: (1) sect. Quinquefoliae subsect. Strobus is monophyletic, (2) sect. Quinquefoliae subsect. Gerardianae is monophyletic and sister to subsect. Strobus, and (3) sect. Parrya is sister to sect. Strobus. Other nuclear markers such as ITS of rDNA (Gernandt et al. 2001) and the LEA-like gene (Syring et al. 2007) also support the above phylogenetic relationship. Therefore, the recent definition of sections and subsections in subgenus Strobus based on cpDNA phylogeny (Gernandt et al. 2005) seems to be highly reliable. Although the ILD test showed that the cpDNA and the two nuclear DNA trees were significantly different from each other except in one case (Table 2), this incongruence was due to topological differences within subsect. Strobus.
In contrast to cpDNA and nrDNAs, the mtDNA tree of the combined dataset showed that the species of both subsect. Strobus and subsect. Gerardianae were split into two groups, GROUP_1 and GROUP_2 (Figs. 1a, b). The constraint of the monophyly of GROUP_1 and _2 on cpDNA and nrDNA datasets resulted in significantly more steps, suggesting that the topological incongruence between mtDNA and the other trees was statistically significant (Table 3). In contrast, the different splitting patterns within subsect. Strobus, which are found in the cpDNA and mtDNA trees, resulted in the same or slightly longer trees in the nrDNA datasets. It is therefore suggested that the major part of the topological anomaly in the mtDNA tree can be attributed to the non-monophyly of subsect. Gerardianae. As for the topological differences within subsect. Strobus, however, it cannot be concluded whether the mtDNA or cpDNA tree depicts the correct phylogenetic relationship of species.
Incongruence within subsect. Strobus
The factors constituting incongruence between mtDNA and the other datasets may be divided into the following two categories: the different splits within subsect. Strobus, and the non-monophyly of subsect. Gerardianae. In order to simplify the explanation of our hypothesis regarding incongruence, we first discuss the former incongruence. Generally, incongruence among different gene trees can be caused by several biological processes: incomplete lineage sorting, orthology and paralogy conflation, and introgressive hybridization. Incomplete lineage sorting is problematic for phylogeny reconstruction in the case that time intervals of successive speciation events are short and not longer than the coalescent time of the genetic marker used (Nei 1987). Willyard et al. (2007) estimated that the divergence point between subsect. Strobus and subsect. Gerardianae occurred approximately 20 million years ago (Mya) based on setting the divergence time of the two subgenera at 85 Mya. Meanwhile, Syring et al. (2007) estimated the time for monophyly of intraspecific nuclear alleles to be more likely than paraphyly (1.67 × 2Ne generations) for three pine species: P. lambertiana (76.3 My), P. discolor (42.6 My) and P. flexilis (5.41 My). Bouillé and Bousquet (2005) also calculated similar values of coalescent time (2Ne generations, 10–18 My) for three Picea species, which have the similar life history traits to pine species. Although the coalescent time of cpDNA and mtDNA is expected to be half that of nuclear genes due to their haploid manner of inheritance (Birky et al. 1983), coalescent times in some pines are still comparable to, or longer than, the whole time span of diversification within subsect. Strobus. In this respect, not only nuclear DNA trees but also both the cpDNA and mtDNA trees could be biased in some part by incomplete lineage sorting.
Even for organellar genes, the problems of orthology and paralogy conflation can arise in cases where there has been intracellular gene transfer (Wendel and Doyle 1998). However, this seems to be unlikely because all nucleotide sequences of mtDNA and cpDNA were successfully determined using a direct sequencing method for PCR products. Additionally, there were no individuals showing a heterozygous-like genotype at the mitochondrial nad1 gene and the chloroplast trnL-trnF region in population genetic analyses of hybrid zones between P. pumila and P. parviflora var. pentaphylla (Senjo et al. 1999; Watano et al. 1996).
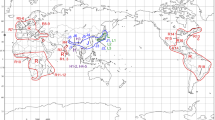
The last possibility, introgressive hybridization, should be carefully considered because there is abundant evidence of cytoplasmic introgression between sympatric or parapatric pine species. However, the detection of past introgression events may be confounded by the incomplete lineage sorting assumed within subsect. Strobus. We propose that a clue to past hybridization events may be found in the geographical distribution patterns of species whose phylogenetic positions are different between the cpDNA and mtDNA trees (Fig. 4). The cpDNA tree (Fig. 2) split species in subsect. Strobus into two groups: P. albicaulis of North America and all Eurasian species except for P. peuce (Eurasian Clade) and then all other species. In contrast, the mtDNA trees (Figs. 1a, b) suggest that all North American species of subsect. Strobus and three Eurasian species, P. pumila, P. koraiensis and P. peuce, are included in GROUP_2 and the other in GROUP_1. In this respect, two Eurasian species (P. pumila and P. koraiensis) and one North American species (P. albicaulis) are characterized by having cpDNA of the Eurasian Clade and mtDNA of GROUP_2, which is a minority combination in both continents. Interestingly, the three species are distributed in the proximity of the Bering Strait (Fig. 4). Beringia, northeastern Siberia and northwestern North America, are thought to have remained ice-free during Quarternary glaciations, and to have played dual roles as both a glacial refugium and a route of colonization across the continents of Eurasia and North America (Hultén 1937). Although this region has long been considered an ice-age refugium for arctic herbs (Abbott and Brochmann 2003), recent studies of palynology and phylogeography have suggested that even boreal trees and shrubs survived in Beringia during the last glacial maximum (Anderson et al. 2006; Brubaker et al. 2005). Based on the geographical distribution of the three species concerned, we propose that the incongruence between the cpDNA and mtDNA trees may have been caused partly by the past cytoplasmic introgression events that occurred in Beringia. The hypothesized introgression may have occurred when species from both continents were trapped in the same glacial refugia in Beringia, or during the colonization from Beringia to new continents after glaciations.

Geographical distribution of the pine species examined (modified from Mirov 1967). As for the species of subsect. Strobus, cpDNA and mtDNA groups of each species are shown by open and filled contours of the distribution range, respectively. Abbreviations of species names: PEU Pinus peuce, CEM P. cembra, SIB P. sibirica, WAL P. wallichiana, ARM P. armandii, FEN P. fenzeliana (cpDNA type is from the sequence of P. kwangtungensis), PAR P. parviflora, KOR P. koraiensis, PUM P. pumila, ALB P. albicaulis, STR P. strobus, FLE P. flexilis, STROBI P. strobiformis
Supporting evidence for the introgression hypothesis would come from observation of ongoing and recent past cytoplasmic introgression events in subsect. Strobus. In eastern Asia, mtDNA of P. pumila (GROUP_2) is permeating into P. parviflora, which originally had GROUP_1 mtDNA (Senjo et al. 1999). On the other hand, in western North America, Liston et al. (2007) has found that cpDNA of northern populations of P. lambertiana—one of the North American species of subsect. Strobus—is replaced by that of P. alibicaulis (Eurasian clade). These findings seem to imply the ongoing spreading process of GROUP_2 mtDNA in Eurasia, and of the Eurasian Clade cpDNA in North America.
A plausible reconstruction of the processes of cytoplasmic introgression events would depend on determining the correct phylogenetic relationship of species, which should be provided by the data independent from both cpDNA and mtDNA. Unfortunately, our two nuclear gene datasets do not contain enough phylogenetic information to determine which split within sect. Strobus is correct (Table 3). However, it should be noted that the LEA-like gene tree (Syring et al. 2007) suggests a similar phylogenetic relationship as that suggested by the mtDNA tree. The LEA-like gene tree resolved five clades (A–E) within subsect. Strobus. Alleles from two Eurasian species (P. pumila and P. koraiensis) both showed species monophyly, and form a clade (clade A) with alleles from five North American species of subsect. Strobus (P. albicaulis, P. strobus, P. monticola, P. lambertiana and P. strobiformis), although bootstrap support of the clade is less than 50%. On the other hand, alleles from the other Eurasian species of subsect. Strobus were grouped into two other clades (D and E), which are composed mostly of alleles from Eurasian species. The phylogenetic affinity of P. pumila and P. koraiensis to the North American species, which is suggested by the mtDNA and LEA-like gene trees, leads to the idea that the two species might be of North American origin. Interestingly, macrofossils (needles and seeds) of P. pumila or a closely related extinct species have been reported from Pliocene sites in Alaska and Canadian Arctic Archipelagos (Matthews and Ovenden 1990). Matthews and Ovenden (1990) also reported needle fossils referred to as subsect. Cembrae sensu Engelmann (1880), which are characterized by resin canals with the medial positions in the needles. Four extant species (P. cembra, P. sibirica, P. koraiensis, P. armandii) have this character and all are confined to Eurasia at present. Macrofossils apparently suggest that late Tertiary flora in arctic North America had contained the five-needle pine species with the morphological characters observed only in extant Eurasian species.
The conflict between the cpDNA and mtDNA datasets suggests that caution should be taken when relying on a single cytoplasmic genomic source for constructing species phylogeny. Definitive answers should be based on data with larger sampling, both geographically (Liston et al. 2007) and through nuclear genomes (Rokas et al. 2003), or the phylogenetic reconstruction method considering explicitly the processes of lineage sorting (Maddison and Knowles 2006).
Non-monophyly of subsect. Gerardianae in mtDNA trees
The non-monophyly of subsect. Gerardianae (P. gerardiana and P. bungeana) is a major point of anomaly in the mtDNA tree. Because the nad1 and nad5 datasets showed different phylogenetic signals concerning the position of P. bungeana (Figs. 1c, d), we examined each PI site in detail (Fig. 5). The combined mtDNA datasets placed P. bungeana at the basal position of GROUP_2 (Figs. 1a, b). If both sequence regions contain the same phylogenetic information, characters supporting a basal branch of GROUP_2 and those supporting monophyly of GROUP_2 species except P. bungeana would be expected to be distributed randomly in both nad1 and nad5 sequences. In fact, however, the former characters are found only in nad1 and the latter only in nad5. The chimeric structure suggests that the mtDNA haplotype of P. bungeana may be caused by intergenic recombination. Given that P. bungeana and P. gerardiana are sister taxa, the nad5 region of P. bungeana, which is similar to that of P. gerardiana, might be original. Therefore, the nad1 region of P. bungeana might have been acquired from a species of subsect. Strobus with GROUP_2 mtDNA, and then become integrated into its mtDNA genome via recombination. Although mtDNA of species in Pinaceae is maternally inherited, examples of occasional paternal leakage has been reported (Wagner et al. 1991). mtDNA recombination prompted by heteroplasmy due to paternal leakage during hybridization has been suggested to explain the mtDNA haplotype variation in the contact zone between two conifer species, Picea mariana and Picea rubens (Jaramillo-Correa and Bousquet 2005). Another possible mechanism may be horizontal mitochondrial gene transfer (Knoop 2004). Horizontal transfer often results in gene duplication or chimeric gene structure (Bergthorsson et al. 2003). Although the examples exemplified so far in seed plants are restricted to the transfer between very distantly related taxa such as Gnetum and asterids (Won and Renner 2003), this possibility cannot be ignored at this stage because there is no record to test reproductive compatibility between P. bungeana and the species of subsect. Strobus. The indel characters of the nad1 intron 2 of P. bungeana are not similar to those of P. pumila, P. koraiensis or P. peuce, which have GROUP_2 mtDNA and are distributed in the Eurasian continent (Table S2). Therefore, the putative donor of the nad1 region to P. bungeana may be an ancestral species of the latter or the indels of P. bungeana may have been modified at the time of recombination.

Chimeric structure of the mtDNA sequence of Pinus bungeana. All phylogenetically informative sites in nad1 intron 2 and nad5 intron 1 are shown. Shading indicates taxa that have a derived character state at given position, which is judged by outgroup comparison. Numbers represent the position in the multiple alignment of the combined dataset
Conclusions
Mitochondrial DNA sequences of the species of Pinus subgenus Strobus were found to have very different phylogenetic information from cpDNA and nuclear DNAs. The incongruence between gene trees may be attributed to the following two factors: the different split patterns within subsect. Strobus, and the non-monophyly of P. bungeana and P. gerardiana of subsect. Gerardianae. The former incongruence was hypothesized to be caused partly by past introgression of cpDNA, mtDNA, or both, between Eurasian and North American species. Phylogeographical considerations suggest that the introgression events occurred at Beringia. The latter incongruence could be explained by the chimeric structure of the mtDNA sequence of P. bungeana, which might have been caused by acquisition of the nad1 segment from species of subsect. Strobus via hybridization or horizontal transfer.
The results reported here show that the phylogenetic relationship within subsect. Strobus is still not settled, even after extensive cpDNA studies. In particular, the phylogenetic affinity of two Far Eastern species (P. pumila and P. koraiensis) to the North American species requires rigorous verification. Beyond phylogenetic concerns, the results of the present study have important implications with regards to cytoplasmic genetic exchanges between species of Eurasian and North American origin.
References
Abbott RJ, Brochmann C (2003) History and evolution of the arctic flora: in the footsteps of Eric Hultén. Mol Ecol 12:99–313
Anderson LL, Hu FS, Nelson DM, Petit RJ, Paige KN (2006) Ice-age endurance: DNA evidence of a white spruce refugium in Alaska. Proc Natl Acad Sci USA 103:12447–12450
Bergthorsson U, Adams KL, Thomason B, Palmer JD (2003) Widespread horizontal transfer of mitochondrial genes in flowering plants. Nature 424:197–201
Birky CW, Maruyama T, Fuerst P (1983) An approach to population and evolutionary genetic theory for genes in mitochondria and chloroplast, and some results. Genetics 103:513–527
Bouillé M, Bousquet J (2005) Trans-species shared polymorphisms at orthologous nuclear gene loci among distant species in the conifer Picea (Pinaceae): implications for the long-term maintenance of genetic diversity in trees. Am J Bot 92:63–73
Brubaker LB, Anderson PM, Edwards ME, Lozhkin AV (2005) Beringia as a glacial refugium for boreal trees and shrubs: new perspectives from mapped pollen data. J Biogeogr 32:833–848
Burban C, Petit RJ (2003) Phylogeography of maritime pine inferred with organelle markers having contrasted inheritance. Mol Ecol 12:1487–1495
Chen J, Tauer CG, Bai G, Huang Y, Payton ME, Holley AG (2004) Bidirectional introgression between Pinus taeda and Pinus echinata: evidence from morphological and molecular data. Can J For Res 34:2508–2516
Critchfield WB (1986) Hybridization and classification of the white pines (Pinus section Strobus). Taxon 35:647–656
Cronn R, Wendel JF (2004) Cryptic trysts, genomic mergers, and plant speciation. New Phytol 161:133–142
Demesure B, Sodzi N, Petit RJ (1995) A set of universal primers for amplification of polymorphic non-coding regions of mitochondrial and chloroplast DNA in plants. Mol Ecol 4:129–131
Dong J, Wagner DB (1993) Taxonomic and population differentiation of mitochondrial diversity in Pinus banksiana and Pinus contorta. Theor Appl Genet 86:573–578
Eckert AJ, Hall BD (2006) Phylogeny, historical biogeography, and patterns of diversification for Pinus (Pinaceae): phylogenetic tests of fossil-based hypotheses. Mol Phylogenet Evol 40:166–182
Engelmann G (1880) Revision of the genus Pinus, and description of Pinus elliottii. Trans Acad Sci St Louis 4:161–190
Farris JS, Källersjö M, Kluge AG, Bult C (1994) Testing significance of incongruence. Cladistics 10:315–319
Garrett PW (1979) Species hybridization in the genus Pinus. USDA Forest Service Research Paper NE-436
Gernandt DS, Liston A, Pinero D (2001) Variation in the nrDNA ITS of Pinus subsection Cembroides: implications for molecular systematic studies of pine species complexes. Mol Phylogenet Evol 21:449–467
Gernandt DS, Lopez GG, Garcia SO, Liston A (2005) Phylogeny and classification of Pinus. Taxon 54:29–45
Gugerli F, Senn J, Anzidei M, Madaghiele A, Buchler U, Sperisen C, Vendramin GG (2001) Chloroplast microsatellites and mitochondrial nad1 intron 2 sequences indicate congruent phylogenetic relationships among Swiss stone pine (Pinus cembra), Siberian stone pine (Pinus sibirica), and Siberian dwarf pine (Pinus pumila). Mol Ecol 10:1489–1497
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hultén E (1937) Outline of the history of Arctic and Boreal biota during the Quaternary period. Cramer, New York
Jaramillo-Correa JP, Bousquet J (2005) Mitochondrial genome recombination in the zone of contact between two hybridizing conifers. Genetics 171:1951–1962
Knoop V (2004) The mitochondrial DNA of land plants: peculiarities in phylogenetic perspective. Curr Genet 46:123–139
Liston A, Robinson WA, Pinero D, Alvarez-Buylla ER (1999) Phylogenetics of Pinus (Pinaceae) based on nuclear ribosomal DNA internal transcribed spacer region sequences. Mol Phylogenet Evol 11:95–109
Liston A, Parker-Defeniks M, Syring JV, Willyard A, Cronn R (2007) Interspecific phylogenetic analysis enhances intraspecific phylogeographical inference: a case study in Pinus lambertiana. Mol Ecol 16:3926–3937
Little EL, Critchfield WB (1969) Subdivision of the genus Pinus (pines), USDA Forest Service, Washington, DC, Miscellaneous Publication Number 1144
Maddison WP, Knowles LL (2006) Inferring phylogeny despite incomplete lineage sorting. Syst Biol 55:21–30
Matos JA, Schaal BA (2000) Chloroplast evolution in the Pinus montezumae complex: a coalescent approach to hybridization. Evolution 54:1218–1233
Matthews JV, Ovenden LE (1990) Late Tertiary plant macrofossils from localities in arctic/subarctic North America: a review of the data. Arctic 43:364–392
Michel F, Umesono K, Ozeki H (1989) Comparative and functional anatomy of group II catalytic introns—a review. Gene 82:5–30
Mirov NT (1967) The genus Pinus. Ronald, New York
Müller K (2005) SeqState: primer design and sequence statistics for phylogenetic DNA datasets. Appl Bioinformatics 4:65–69
Neale DB, Sederoff RR (1989) Paternal inheritance of chloroplast DNA and maternal inheritance of mitochondrial DNA in loblolly pine. Theor Appl Genet 77:212–216
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Nylander JAA (2004) MrModeltest v2. Program distributed by the author. Evolutionary Biology Centre, Uppsala University
Price RA, Liston A, Strauss SH (1998) Phylogeny and systematic of Pinus. In: Richardson DM (ed) Ecology and biogeography of Pinus. Cambridge University Press, Cambridge, pp 49–68
Richardson BA, Brunsfeld SJ, Klopfenstein NB (2002) DNA from bird-dispersed seed and wind-disseminated pollen provides insights into postglacial colonization and population genetic structure of whitebark pine (Pinus albicaulis). Mol Ecol 11:215–227
Rieseberg LH, Soltis DE (1991) Phylogenetic consequences of cytoplasmic gene flow in plants. Evol Trend Plant 5:65–84
Rokas A, Williams BL, King N, Carroll SB (2003) Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 425:798–804
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Senjo M, Kimura K, Watano Y, Ueda K, Shimizu T (1999) Extensive mitochondrial introgression from Pinus pumila to P. parviflora var. pentaphylla (Pinaceae). J Plant Res 112:97–105
Simmons MP, Ochoterena H (2000) Gaps as characters in sequence based phylogenetic analyses. Syst Biol 49:369–381
Suyama Y, Yoshimaru H, Tsumura Y (2000) Molecular phylogenetic position of Japanese Abies (Pinaceae) based on chloroplast DNA sequences. Mol Phylogenet Evol 16:271–277
Swofford DL (2003) PAUP*. Phylogenetic analysis using parsimony (*and other methods). version 4. Sinauer Associates, Sunderland
Syring J, Willyard A, Cronn R, Liston A (2005) Evolutionary relationships among Pinus (Pinaceae) subsections inferred from multiple low-copy nuclear loci. Am J Bot 92:2086–2100
Syring J, Farrell K, Businsky R, Cronn R, Liston A (2007) Widespread genealogical nonmonophyly in species of Pinus Subgenus Strobus. Syst Biol 56:163–181
Tani N, Maruyama K, Tomaru N, Uchida K, Araki M, Tsumura Y, Yoshimaru H, Ohba K (2003) Genetic diversity of nuclear and mitochondrial genomes in Pinus parviflora Sieb. & Zucc. (Pinaceae) populations. Heredity 91:510–518
Temesgen B, Brown GR, Harry DE, Kinlaw CS, Sewell MM, Neale DB (2001) Genetic mapping of expressed sequence tag polymorphism (ESPT) markers in loblolly pine (Pinus taeda L.). Theor Appl Genet 102:664–675
Templeton AR (1983) Phylogenetic inference from restriction endonuclease cleavage site maps with particular reference to the evolution of humans and the apes. Evolution 37:221–244
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Wagner DB, Furnier GR, Saghai-Maroof MA, Williams SM, Dancik BP, Allard RW (1987) Chloroplast DNA polymorphisms in lodgepole and jack pines and their hybrids. Proc Natl Acad Sci USA 84:2097–2100
Wagner DB, Dong J, Carlson MR, Yanchuk AD (1991) Paternal leakage of mitochondrial DNA in Pinus. Theor Appl Genet 82:510–514
Wang XR, Tsumura Y, Yoshimaru H, Nagasaka K, Szmidt AE (1999) Phylogenetic relationships of Eurasian pines (Pinus, Pinaceae) based on chloroplast rbcL, matK, rpl20-rps18 spacer, and trnV intron sequences. Am J Bot 86:1742–1753
Wang XQ, Tank DC, Sang T (2000) Phylogeny and divergence times in Pinaceae: evidence from three genomes. Mol Biol Evol 17:773–781
Watano Y, Imazu M, Shimizu T (1995) Chloroplast DNA typing by PCR-SSCP in the Pinus pumila-P. parviflora var. pentaphylla Complex (Pinaceae). J Plant Res 108:493–499
Watano Y, Imazu M, Shimizu T (1996) Spatial distribution of cpDNA and mtDNA haplotypes in a hybrid zone between Pinus pumila and P. parviflora var. pentaphylla (Pinaceae). J Plant Res 109:403–408
Watano Y, Kanai A, Tani N (2004) Genetic structure of hybrid zones between Pinus pumila and P. parviflora var. pentaphylla (Pinaceae) revealed by molecular hybrid index analysis. Am J Bot 91:65–72
Wendel JF, Doyle JJ (1998) Phylogenetic incongruence: windows into genome history and molecular evolution. In: Soltis DE, Soltis PS, Doyle JJ (eds) Molecular systematics of plants II DNA sequencing. Kluwer, Dordrecht, pp 265–296
Willyard A, Syring J, Gernandt DS, Liston A, Cronn R (2007) Fossil calibration of molecular divergence infers a moderate mutation rate and recent radiations for Pinus. Mol Biol Evol 24:90–101
Won H, Renner SS (2003) Horizontal gene transfer from flowering plants to Gnetum. Proc Natl Acad Sci USA 100:10824–10829
Acknowledgments
The authors thank Dr. Yoshihiko Tsumura for providing genomic DNA samples of Pinus sibirica; the Royal Botanic Garden Edinburgh for providing dried leaves of P. albicaulis, P. cembra, P. koraiensis and P. peuce; the Field Science Education and Research Center of Kyoto University for providing leaf samples of many other pine species. This study was partly supported by Japan Society for the Promotion of Science, Grant-in-Aid for Scientific Research (B), No. 14340265 and No. 18370033.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tsutsui, K., Suwa, A., Sawada, K. et al. Incongruence among mitochondrial, chloroplast and nuclear gene trees in Pinus subgenus Strobus (Pinaceae). J Plant Res 122, 509–521 (2009). https://doi.org/10.1007/s10265-009-0246-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10265-009-0246-4