
Abstract
Despite recent data that suggest that the overall performance of drug-eluting stents (DES) is superior to that of bare-metal stents, the long-term safety and efficacy of DES remain controversial. The risk of late stent thrombosis associated with the use of DES has also motivated the development of a new and promising treatment option in recent years, namely drug-coated balloons (DCB). Contrary to DES where the drug of choice is typically sirolimus and its derivatives, DCB use paclitaxel since the use of sirolimus does not appear to lead to satisfactory results. Since both sirolimus and paclitaxel are highly lipophilic drugs with similar transport properties, the reason for the success of paclitaxel but not sirolimus in DCB remains unclear. Computational models of the transport of drugs eluted from DES or DCB within the arterial wall promise to enhance our understanding of the performance of these devices. The present study develops a computational model of the transport of the two drugs paclitaxel and sirolimus eluted from DES in the arterial wall. The model takes into account the multilayered structure of the arterial wall and incorporates a reversible binding model to describe drug interactions with the constituents of the arterial wall. The present results demonstrate that the transport of paclitaxel in the arterial wall is dominated by convection while the transport of sirolimus is dominated by the binding process. These marked differences suggest that drug release kinetics of DES should be tailored to the type of drug used.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Despite the recent data of the Swedish Coronary Angiography and Angioplasty Registry (SCAAR) study (Sarno et al. 2012) that suggest that the overall performance of drug-eluting stents (DES) is superior to that of bare-metal stents (BMS), the long-term safety and efficacy of DES remain controversial. The primary concern stems from the risk of late and very late stent thrombosis, thought to be principally attributable to delayed endothelial healing at the site of stent implantation. In an attempt to avoid the complications associated with DES, an emerging technology is the use of drug-coated balloons (DCB), either with or without subsequent stent implantation (Gray and Granada 2010). Interestingly, all DCB devices currently under development use paclitaxel as the therapeutic agent of choice. This is in contrast to developments in the latest-generation DES, where manufacturers have for the most part opted for sirolimus or its derivatives as the drug of choice. DCB and DES have fundamentally different strategies for delivering drugs to the arterial wall: DCB deliver a very high drug dose (\(200{-}300\,\upmu \hbox {g}/\hbox {cm}^{2}\)) in a very short time, whereas DES deliver a considerably lower dose (\(100\,\upmu \hbox {g}/\hbox {cm}^{2}\)) over a period of several weeks.
Another important finding of the SCAAR study is that second-generation DES (such as Medtronic’s Endeavor Resolute stent or Abbott’s Xience V stent), which have undergone major design changes in their geometry, drug composition, and the composition of the polymeric coating within which the drug is embedded relative to first-generation DES, exhibit clearly superior performance. Most investigations to date of the effect of stent design on stent performance have focused on the role of detailed geometric features (Balakrishnan et al. 2005; LaDisa 2004; Seo et al. 2005) or of DES polymeric coating (Hara et al. 2006). The role of the drug elution process itself (Balakrishnan et al. 2007) and its coupling to the specific drug used (Tzafriri et al. 2012) have received less attention and have thus far been limited to sirolimus.
Computational models of the transport of drugs eluted from DES or DCB within the arterial wall promise to significantly enhance our understanding of the performance of these devices. A number of different modeling approaches have been proposed ranging from relatively simple models that assume either a low drug diffusivity in the arterial wall (Mongrain et al. 2007) or a constant partition of bound and free drug Zunino (2004) to more sophisticated models where drug interactions with the arterial wall are described by a second-order reversible reaction (Tzafriri et al. 2009). Multidimensional models (Mongrain et al. 2007; Borghi et al. 2008), with the exception of the 3D model by Feenstra and Taylor (2009), approximate the arterial wall as a single homogeneous layer and often neglect convective drug transport within the wall. Only 1D studies (Pontrelli and Monte 2010; McGinty et al. 2011) have thus far included the layered structure of the arterial wall into their models for describing the elution of drugs from stents. The diversity of modeling approaches, the different assumptions on which the models are based, and the limited experimental validation of the models render it difficult to compare the predictions of the different models and to make general conclusions. In some cases, different models can even lead to contradictory predictions: for example, the calculations by Vairo et al. (2010) indicated insensitivity of the drug concentration profile in the arterial wall to lumenal blood flow, while the work by Balakrishnan et al. (2005) reached the opposite conclusion.
In an effort to better understand the validity and applicability of different model assumptions, the present work develops a computational model of the transport of the two drugs paclitaxel and sirolimus eluted from DES in the arterial wall. The model takes into account the multilayered structure of the arterial wall and incorporates a reversible binding model to describe drug interactions with the constituents of the arterial wall. The effects of assuming a one-layer model for the arterial wall or equilibrium reaction conditions are explored. Finally, the model is used to directly compare the coupling between the drug release kinetics from the stent and the drug dynamics in the arterial wall for the two DES drugs studied.
2 Materials and methods
2.1 Model geometry
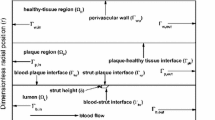
A 2D axisymmetric geometry is used in all simulations. We consider a straight arterial segment within which a model DES represented by ten circular struts is deployed (Fig. 1).

A Model geometry with stent strut diameter \(d_\mathrm{s}\!=\!\text {metallic strut diameter}\!+\!2\cdot \text {polymer thickness}\!=\!0.15\,\hbox {mm}\!+\!2\times 0.05\,\hbox {mm}\!=\!0.25\,\hbox {mm}\). All dimensions are in millimeters. Geometry not drawn to scale. B Computational mesh: left: close-up on struts 4 through 6. Right: detail of the intersection of polymer, lumen, SES, and media with indication of boundary layer elements
Stent strut size, polymer thickness, and stent interstrut spacing are adapted from the geometry introduced by Mongrain et al. (2005) and recently also used by Vairo et al. (2010). Due to the reduction in the number of struts relative to those studies (10 struts instead of 15), our geometry is considered to represent a smaller lesion (7 mm). We have verified that this has no significant impact on the parameters of interest. Embedment of the stent struts in the arterial wall is assumed to be 50 %. A sensitivity study investigating the effect of strut embedment in the 20–80 % range revealed that while the magnitude of drug concentration in the arterial wall changes, the overall behavior and the conclusions drawn are not altered. We also define a therapeutic domain that extends three interstrut spacings both upstream and downstream of the stent (Fig. 1) and that we consider to be the target zone for drug delivery. All computed variables are for this therapeutic domain.
2.2 Physical model
The mathematical framework for the description of the arterial wall is based on the two-layer model presented by Formaggia et al. (2009). We apply this model to the elution of the two small hydrophobic drugs paclitaxel and sirolimus from DES and complement it with a recently developed reaction model in the arterial wall (Tzafriri et al. 2009). Wherever possible, the model input parameters are derived from experimental data available in the literature.
The arterial wall is assumed to be rigid and is modeled as a two-layered structure with the subendothelial space (SES) and the media defined as distinct domains, while the endothelium and internal elastic lamina (IEL) are treated as interfacial matching conditions. The adventitia is not modeled as a distinct layer but rather as a boundary condition at the outer surface of the media. Because stent deployment is thought to lead to near-complete denudation of the endothelium at the deployment site, we assume in the modeling a baseline configuration where the endothelium is absent within the stented portion of the vessel but is intact otherwise.
Drug transport is assumed to occur in the following four different domains: the arterial lumen, the stent polymer coating, the SES, and the media. Different physical phenomena dominate transport in each of these domains. In the polymer coating, we assume drug transport to be purely diffusive. In the lumen, drug is transported via both convection and diffusion. Each of the two layers of the arterial wall, i.e., the SES and the media, is modeled as a porous medium with its distinct homogenized (but in some cases anisotropic) properties. The SES is assumed to be devoid of all cells, so that drug transport within this layer occurs by convection and diffusion. In the media, three different processes govern drug concentration: convective and diffusive transport within the pore space, specific binding of the drug to smooth muscle cells (SMCs), and nonspecific binding of the drug to the extracellular matrix accounted for through hindered diffusion.
2.3 Governing equations and boundary conditions
Unless stated otherwise, variables and equations are presented in non-dimensional form. Dimensional quantities are denoted by a tilde, and the reference values used in the non-dimensionalization are provided in Table 1.
2.3.1 Momentum transport
In the arterial lumen, blood flow is assumed to be steady and is thus governed by the time-independent Navier–Stokes equations:
where \(\varvec{u}_\mathrm{l}\) and \(p_\mathrm{l}\), respectively, denote the velocity and pressure in the lumen. Blood is assumed to be a Newtonian fluid, and a lumenal Reynolds number of \( Re _\mathrm{l}={\tilde{\bar{u}} \tilde{d}_\mathrm{l} \tilde{\rho }}/{\tilde{\mu }_\mathrm{b}}=400\) is considered based on blood velocity magnitudes typically encountered in coronary arteries.
The SES and the media are considered as porous layers, and flow within these layers is assumed to be governed by Darcy’s law:
where the subscript \(j\) denotes either the SES or the media, \(\varvec{u}_j\) is the average fluid velocity in the total volume (matrix plus pores), and \(\mu _{\mathrm{p}}\) and \(P_{\mathrm{D,}j}\) are the non-dimensional dynamic viscosity and Darcy permeability, respectively. In the polymeric coating of the stent, the flow is assumed absent and thus, drug transport occurs solely by diffusion.
The following boundary conditions apply: At the inlet of the lumen region, a laminar Poiseuille velocity profile is prescribed:
while at the outlet, the pressure is fixed at the physiological excess pressure value of \(\tilde{p}_\mathrm{out}= 100\,\hbox {mmHg}\). The top boundary of the lumenal domain is the symmetry axis of the problem. The endothelium and IEL are approximated as semipermeable membranes with the endothelium coupling flow in the lumen to that in the SES and the IEL serving as an interfacial matching condition between the SES and the media. To describe these membranes, we use the Kedem–Katchalsky equation (Kedem and Katchalsky 1958) governing the fluid flux \(J_\mathrm{v}\) across these membranes. Due to the very small size of the drug molecules (\(\approx \)1 nm, Hilder and Hill 2008) compared with the junction size of the endothelium (leaky junction size \(\approx \)20 nm) or the pore size of the IEL (\(\approx \)150 nm), the osmotic reflection coefficient is very small (\(\propto \left( {r_\mathrm{mol}}/{r_\mathrm{pore}}\right) ^2\)), and the flux simplifies to (Formaggia et al. 2009)
where \(L_{\mathrm{p,}j}\) is the hydraulic conductivity of the respective membrane. In the central region where endothelial cells do not hinder the fluid flow across the arterial wall, the matching condition simplifies to continuity of radial velocity (\(u_\mathrm{l} = u_\mathrm{ses}\)). In all cases, the axial velocity at the lumen/arterial wall interface is set to zero (\(w_\mathrm{l} = 0\)). A zero axial pressure gradient is assumed normal to the upstream and downstream boundaries of the arterial wall layers, resulting in no fluid flux across these boundaries and imposing a strictly radial flow field at these boundaries. This assumption is strictly valid in an axially homogeneous arterial wall; the upstream and downstream boundaries are sufficiently far from the therapeutic domain so that these boundary conditions have no effect on the results. On the adventitial boundary (outer boundary of the arterial wall), an excess pressure of \(\tilde{p}_\mathrm{eel}= 30\,\hbox {mmHg}\) is assumed.
2.3.2 Drug transport
Drug transport in the lumen is governed by the unsteady convection–diffusion equation:
Equation (6) describes the concentration \(c_{\alpha ,j}\) of a drug transported in the liquid phase (intrinsic drug concentration) with a non-dimensional diffusion coefficient \(D_{\alpha ,j}\) at a reference Peclet number \( Pe _0\) (see Table 1). To describe mass transport in the porous arterial wall, averaging is required. Neglecting dispersion, superficial averaging (averaging over both phases of the porous medium containing the pore space and the solid tissue space) as described by Whitaker (2010) yields the following macroscopic transport equation in the arterial wall (henceforth simply referred to as transport equation):
where the reaction term \(R_j\) accounts for drug flux to and from the tissue corresponding to the binding and unbinding of drug molecules to their binding sites in the arterial wall. The two parameters that arise in the averaging process, the non-dimensional lag coefficient \(\varLambda _j\) and the effective non-dimensional diffusivity \({\mathsf {D}}_j\), will be discussed later. For a pure fluid (porosity \(\varepsilon =1\)), \(\varLambda _j=1\) and the effective diffusivity equals the diffusion coefficient \(D_{\alpha ,j}\) in the fluid (in our case, \(D_{\alpha \mathrm{,l}}=1\) and \(\varvec{u}_{\alpha \mathrm{,l}}\) is the velocity calculated from the Navier–Stokes equations in the lumen). In this case, the liquid concentration \(c_{\alpha ,j}\) equals the superficial average concentration \(c_j\).
A general way of expressing the reaction term \(R_j\) is by:
which describes reaction as the negative of the rate of creation of bound drug concentration \(b\) as the drug binds to the porous material. The factor \(f_\mathrm{cb}\) is defined as \({\tilde{c}_0}/{\tilde{b}_\mathrm{m}}\), the ratio of the initial concentration \(\tilde{c}_0\) in the stent polymer to the maximum binding site density \(\tilde{b}_\mathrm{max}\). In recent literature, two common ways of modeling drug uptake by arterial tissue can be found:
-
1.
Equilibrium model: a simplified approach to model drug uptake assuming that the bound and free states are in a constant equilibrium (Zunino 2004; Vairo et al. 2010). This assumption is valid when the binding and unbinding processes are fast compared with convection and diffusion and leads to:
$$\begin{aligned} R_j = \left( \kappa -1 \right) \frac{\partial c_j}{\partial t}, \end{aligned}$$(9)where \(\kappa \) is the total binding coefficient.
-
2.
Dynamic model: a second-order model that describes a saturating reversible binding process (Tzafriri et al. 2009) treating bound drug as a dynamic variable:
$$\begin{aligned} \frac{1}{f_\mathrm{cb}} \frac{\partial b}{\partial t} = Da _{0} \left( c_j \left( 1-b\right) -\frac{1}{f_\mathrm{cb} B_\mathrm{p}} b \right) \end{aligned}$$(10)
with the reference Damköhler number \( Da _{0}\) and the binding potential \(B_\mathrm{p}\). The baseline model in the present work uses this dynamic model to describe drug interaction with cells of the arterial wall; however, we also explore the ramifications of an equilibrium model assumption for the predictions of drug transport from DES.
The transport equations are subject to the following boundary conditions: zero drug concentration at the inlet boundary in the lumen. Because convection is dominant in the lumen, we set the outlet boundary condition to: \(\varDelta c=0\). At the upstream and downstream boundaries of the arterial wall layers, a zero normal concentration gradient is prescribed. At the adventitial boundary, we investigated the following three different boundary conditions:
-
1.
\(c_\mathrm{m}=0\): considering the adventitia as a perfect sink.
-
2.
\(-\nabla \left( {\mathsf {D}}_\mathrm{m}\nabla c_\mathrm{m} \right) =0\): assuming drug transport across the adventitia to be purely convective.
-
3.
\(J_\mathrm{s,eel}=P_\mathrm{eel} c_{\alpha \mathrm{,m}} + \bar{c}_{\alpha \mathrm{,eel}} J_\mathrm{v,eel}\): modeling the external elastic lamina (EEL) as a Kedem–Katchalsky membrane assuming the drug concentration in the adventitia to be negligible.
We have verified that the choice of boundary condition has no significant effect on the parameters of interest. The baseline model uses the third option since it is considered the closest to the physiological situation.
At both the lumen–arterial wall interface and the SES–media interface, the Kedem–Katchalsky formulation is used to describe the concentration discontinuity across the thin endothelium and IEL:
where \(P_{j}\) is the permeability of the respective interface and the local average liquid concentration \(\bar{c}_{\alpha ,j}\) is calculated as the weighted average concentration of the layer (for more details see “Appendix 2”).
At the outer boundary of the stent polymer, drug flux continuity is prescribed:
Drug flux across the stent strut surface is zero, and the polymer is the only initial drug reservoir yielding the following initial condition:
-
\(\tilde{c}\left( \tilde{t}=0 \right) =\tilde{c}_0= 100\,\hbox {mol}\,\hbox {m}^{-3}\) in the polymer.
-
\(c=0\) elsewhere.
This initial concentration is representative of a high-dose stent (Radeleff et al. 2010).
2.4 Determination of physiological parameters
Special attention was paid to the choice of the physiological parameters of the model. We use experimental data wherever available, complemented with values from fiber matrix and pore theory (Formaggia et al. 2009 and references therein) when necessary. The solely tissue-dependent parameters, namely the hydraulic conductivity \(\tilde{L}_\mathrm{p}\), the porosity \(\varepsilon \), the Darcy permeability \(\tilde{P}_\mathrm{D}\), and the properties of blood and plasma, are well documented in the literature. All values with their respective sources are summarized in Table 2. Fluid density is assumed to be the same for plasma and whole blood with \(\tilde{\rho }=1{,}060\,\hbox {kg}\,\hbox {m}^{-3}\). The hydraulic conductivities of the endothelium and IEL were calculated from \(L_{\mathrm{p,}j}={P_{\mathrm{D,}j}}/{\left( \mu _\mathrm{p} L_j \right) }\) following the approach of Ai and Vafai (2006) and Prosi et al. (2005). The fiber matrix porosity and effective porosity of the media were, respectively, set to \(\varepsilon _\mathrm{f,m} = 0.45\) and \(\varepsilon _\mathrm{m} = 0.25\), yielding a SMC fraction in the media of \(\chi _\mathrm{SMC} = 0.44\), which agrees well with previous data (Karner et al. 2001; Formaggia et al. 2009) (see “Appendix 5” for a detailed definition of the different porosities).
The effective diffusivities for the different layers \({\mathsf {D}}_j\), the lag coefficients \(\varLambda _j\), the membrane permeabilities \(P_j\), and the sieving coefficients \(s_j\) depend on the interplay of the drug with the surrounding tissue. Here, we will mainly focus on the diffusivities used in the model. The calculation of the remaining parameters can be found in “Appendix 6.”
In the present study, we will consider the two DES drugs paclitaxel and sirolimus. Paclitaxel is a small hydrophobic agent with a molecular weight of \(\tilde{M}_\mathrm{PAX}=853.9\;\mathrm{Dalton}\) (Levin et al. 2004) and an average radius of \(\tilde{r}_\mathrm{PAX}=1.2\,\hbox {nm}\) (Hilder and Hill 2008). Diffusion of paclitaxel in blood is reduced due to nonspecific binding to blood proteins; therefore, the free diffusivity in plasma for paclitaxel is taken as the measured value in calf serum as determined by Lovich et al. (2001) \(\tilde{D}_\mathrm{free,p,PAX}=20.3\times 10^{-12}\,\hbox {m}^{2}\,\mathrm{s}^{-1}\). Using this value, the free diffusivity in whole blood is determined using the Stokes–Einstein equation as:
The effective diffusivity \({\mathsf {D}}_j\) in Eq. (7) is a lumped parameter that arises from the averaging process of the diffusive term which accounts for the reduced transport of the solute on its tortuous path through the fibrous structure of the extracellular matrix. Apart from this passive effect, it can also account for an active reduction due to nonspecific binding to the tissue of the arterial wall. No experimental work on the effective diffusivity in the SES is known to the authors. Given the assumptions of our model, we will consider the effective diffusion coefficient to be a simple scaling of the free diffusivity \(\tilde{D}_\mathrm{free}\) by a factor that depends solely on material properties (Whitaker 1986). We will thus approximate the effective diffusivity as
where \(\tilde{r}_\mathrm{mol}\) is the average radius of the drug molecule and \(\tilde{r}_\mathrm{f}\) is the average radius of the fibers of the extracellular matrix. Levin et al. (2004) determined experimentally anisotropic effective bulk diffusion coefficients for paclitaxel, corresponding to the diffusivities in the media of the arterial wall. These measurements suggest a very high anisotropy between radial and axial diffusivities with \({D_z}/{D_r}\approx 1{,}000\). The work conducted by Hwang et al. (2001) suggests an anisotropy between radial and circumferential diffusivities in the media of \({D^\varphi }/{D^r} \approx 10\). The structure of the media as investigated by O’Connell et al. (2008) suggests that the circumferential and axial diffusion coefficients are of the same order of magnitude, whereas the diffusion coefficient in the radial direction is significantly smaller. Therefore, in our baseline model, we assume the axial and circumferential effective diffusivities to be of a comparable order of magnitude (Weinbaum et al. 1985) and \({D^z}/{D^r}=10\). Because the importance of anisotropic diffusion has been highlighted by Hwang and Edelman (2002), we will explicitly investigate the effect of different anisotropies. A summary of all coefficients for the transport of paclitaxel can be found in Table 3.
Similar to paclitaxel, sirolimus is a small hydrophobic drug that binds to the FKBP12 receptor of SMCs and endothelial cells. To calculate the transport properties of sirolimus, we applied the same procedure as described for paclitaxel. Due to the slightly larger molecular weight of \(\tilde{M}_\mathrm{SIR} = 914.2\;\mathrm{Dalton}\) (Levin et al. 2004), the Stokes–Einstein radius and blood plasma diffusivity need to be adjusted (details in “Appendix 7”). All transport coefficients for sirolimus are summarized in Table 3.
It can be readily seen that the diffusivities of paclitaxel and sirolimus are not very different. Rather, the primary difference between the two drugs stems from differences in their reaction kinetics. More specifically, sirolimus binds and unbinds significantly more rapidly than paclitaxel as seen in Table 4. The diffusion coefficient in the stent polymer coating is the result of an interplay of several factors including the polymer matrix, the drug, and the stent platform release kinetics. Balakrishnan et al. (2007) showed that simple diffusion is appropriate to model this complex process. Reported values for diffusion coefficients range from \(\tilde{D}_\mathrm{c}=1\times 10^{-13}\,\hbox {m}^{2}\,\hbox {s}^{-1}\) (Vairo et al. 2010) down to \(\tilde{D}_\mathrm{c}=1.5\times \,10^{-17}\,\hbox {m}^{2}\,\hbox {s}^{-1}\), representing the range from fast- to slow-release kinetics. The baseline configuration in this study assumes a drug diffusivity in the polymer at the fast-release end of this range (\(\tilde{D}_\mathrm{c}=1\times \,10^{-13}\,\hbox {m}^{2}\,\hbox {s}^{-1}\)).
2.4.1 Timescales and dimensionless quantities for drug transport
In order to better interpret the results, it is useful to analyze the timescales introduced by the dynamic reaction model (Eq. 10) and compare them to the other timescales in the transport equation (Eq. 7). Considering the dimensional form of Eq. (10):
the first term on the right-hand side (RHS) represents drug binding to sites in the arterial wall, which requires the presence of both free drug and free binding sites. The binding rate is additionally controlled by the binding rate coefficient \(\tilde{k}_\mathrm{f}\) and the porosity \(\varepsilon _j\) of the tissue. Thus, the characteristic timescale for drug binding is:
The second term on the RHS of Eq. (15) describes the drug unbinding due to the assumed reversible nature of the reaction. For simplicity, we denote \({\tilde{k}_\mathrm{r}}/{\left( 1-\varepsilon _j \right) }\) as \(\tilde{k}^*_\mathrm{r}\), which is the drug unbinding rate constant corrected for the solid tissue fraction. The typical timescale characterizing unbinding is:
The ratio of the (corrected) unbinding rate constant \(\tilde{K}_\mathrm{d}={\tilde{k}^*_\mathrm{r}}/{\tilde{k}_\mathrm{f}}\) to the binding rate coefficient is the equilibrium dissociation constant (Tzafriri et al. 2009) and characterizes the overall affinity of the drug to its binding sites with low values corresponding to high affinity.
We would now like to compare the timescales of drug binding and unbinding to those of drug convection and diffusion. The typical flow velocity in the arterial wall is very different from that in the lumen. The characteristic radial convective flow velocity in the arterial wall can be written as:
with
with \(j\) summed over the endothelium, SES, and media. With this we can now calculate the typical timescale for convection in the media as
and for diffusion
To weigh the relative importance of these various processes, we form the following three dimensionless quantities:
-
Peclet number \( Pe \): ratio of diffusion timescale to convection timescale,
-
Damköhler number \( Da \): ratio of diffusion timescale to binding timescale,
-
and the ratio of the former two dimensionless quantities \(\frac{ Da }{ Pe }\): ratio of convection timescale to binding timescale.
The resulting computed timescales and dimensionless quantities, summarized in Table 5 (the different magnitude of the Damköhler number compared to Tzafriri et al. (2009) stems from the adapted length scale (\(\tilde{L}_\mathrm{m}\)) and diffusivity (\(\tilde{D}^r_{\mathrm{m}}\)) used), indicate that due to differences in their reaction properties, paclitaxel and sirolimus concentration profiles in the arterial wall may be very different despite the fact that both drugs have largely similar transport properties. For both drugs, convection and binding clearly dominate diffusion. The major difference in the two drugs lies in the relative importance of convection compared to reaction: Sirolimus’ binding rate is so high that it even dominates the convective transport (\(\frac{ Da }{ Pe }\gg 1\)), whereas paclitaxel is more sensitive to convection (\(\frac{ Da }{ Pe }<1\)). This table reveals an additional important timescale for the transport problem, namely the release time from the stent, which approximates the time over which the stent effectively releases its drug load from the polymer coating. Thus, for the baseline stent, fresh drug supply only lasts for several hours.
The Damköhler number and the ratio of the Damköhler and Peclet number defined above compare the initial maximum reaction rate to the rates of diffusion and convection, respectively. This is adequate when considering a first-order reaction process but is limited in its informative value when the reaction is second order and is reversible. To get a more comprehensive view of the binding and unbinding process, we define an integral time-dependent Damköhler number which compares the averaged magnitude of the reaction term to that of the convection term in the media at each time point:
2.4.2 One-layer approximation
Due to the small thickness of the intima compared to the media, it is tempting to simplify the problem and represent the intima as a single Kedem–Katchalsky matching condition. Consequently, the arterial wall would consist of a single porous layer representing the media. We wanted to probe the validity of this assumption and to compare the results of the one-layer model to those of our baseline two-layer model. To generate the equivalent effective hydraulic conductivities for the matching boundary condition with and without endothelial coverage, we reinterpreted the different layers involved (endothelium, SES, and IEL) as flow resistances in series (Prosi et al. 2005) and used the total resistance to obtain an effective hydraulic conductivity as:
For the effective parameters for solute transport, we choose the same values as for the endothelium in the non-denuded case and those of the IEL in the denuded case, since these correspond to the largest resistances to transport.
2.5 Numerical methods
The governing equations are discretized by means of the finite element method (FEM) using the commercial software package COMSOL Multiphysics 4.3 (COMSOL AB, Burlington, MA, USA). The following types of elements are used:
-
Momentum transport: third-order Lagrangian elements for the velocity and second-order Lagrangian for the pressure (P3-P2)
-
Drug transport: second-order Lagrangian elements
-
Reaction equation: second-order discontinuous Lagrangian elements
The tolerance threshold for the relative error of the solution (relative tolerance) of the momentum equations was set to \(10^{-9}\). An analysis of the transport problem showed no change of the solution below a combination of relative tolerance \(10^{-3}\) and absolute tolerance \(10^{-4}\). The computational domain of the momentum equations was extended beyond that of the drug transport to ensure insensitivity of the results to the inflow and outflow boundary conditions. The time-advancing scheme is a backward differentiation formula (BDF) with variable order and time step size (Hindmarsh and Brown 2005). The maximum time step size is restricted to 1 h. Reducing the maximum time step to \({1}/{8}\;\hbox {h}\) did not change the solution, validating our choice for the maximum step size.

Baseline simulation (paclitaxel). A Normalized concentration (NC) distribution in the media and the SES at three different time points: \(\tilde{t}=50\,\hbox {min}, \tilde{t}=1\,\hbox {day}\), and \(\tilde{t}=7\,\hbox {day}\) post-stenting. B Temporal evolution of the normalized mean concentration (NMC) in the SES and the media
The minimum mesh element size is set to two times the concentration boundary layer thickness \(\tilde{\delta }_\mathrm{c}\) as estimated by \(\tilde{\delta }_\mathrm{c} \approx 2 \sqrt{3} \tilde{d}_\mathrm{s} Re _\mathrm{d}^{-{1}/{2}} Sc _\mathrm{b}^{-{1}/{3}}\) using the Reynolds number \( Re _\mathrm{d}\) based on the (hydrodynamic) stent diameter \(\tilde{d}_\mathrm{s}\) and the Schmidt number in blood \( Sc _\mathrm{b}\) (Vairo et al. 2010). We note that \( Re _\mathrm{d}= Re _0\) in this particular case, since the reference length and velocity scales used to non-dimensionalize the governing equations are the same as the length and velocity scales used to compute \( Re _\mathrm{d}\). The mixed triangular and quadrilateral mesh (Fig. 1) is enhanced with boundary layer elements (BLE) at the interface between lumenal flow and arterial wall and at the interface of the stent polymer coating with the arterial wall. To smoothen the sharp initial condition from the stent polymer to the surrounding domain, the inner boundary of the triangular polymer mesh is enhanced with BLEs and the initial condition itself transitions from \(c\left( t=0 \right) =1\) to \(c\left( t=0 \right) =0\) using an infinitely differentiable step function. The classical approach of a mesh independence study AIAA (1998) determined the number of elements in the lumen, the SES, and the polymer. We consecutively increased the number of mesh elements in each of these layers by a factor of 1.5–2 until the time evolution of the average concentration in the SES and the polymer showed a relative difference of \(<\)1 % from one mesh iteration to another. Similarly, we used the average wall shear stress along the lumen–wall interface and the flow profile downstream of the stent as the test quantities to verify grid independence in the lumen. In the media, however, the maximum cell size was limited by the occurrence of spurious oscillations in the solution. This resulted in an overall very fine mesh with approximately 290,000 elements. Computation time for the baseline simulation performed on \(4\) cores of an Intel® Xeon® CPU X5680@ 3.33 GHz processor is about 1 h.
3 Results
3.1 Baseline model
To capture the global predictions of the baseline model, we compute the spatially averaged normalized mean concentration (NMC) of the eluted drug in each of the layers \(j\) of the arterial wall. The NMC at any instant in time is defined as follows:
In the case of the one-layer model (OLM), the theoretical subendothelial space (SES) NMC can be calculated from:
Figure 2 shows simulation results for the baseline model eluting paclitaxel from the stent coating at the fastest rate of the presented range. Representative of the entire stent, Fig. 2a compares the total drug concentration (i.e., bound drug \(b\) plus free drug \(c\)) distribution in the upstream region of the therapeutic domain up to the first two stent struts. The three time points after stent implantation shown are: \(\tilde{t}=50\,\hbox {min}\), which corresponds to the time of maximum NMC in the media; \(\tilde{t}=1\;\text {day}\); and \(\tilde{t}=7\,\hbox {day}\) post-implantation. At \(\tilde{t}=50\,\hbox {min}\), the drug has already completely invaded the arterial wall. From the second strut onward, a symmetric drug distribution pattern surrounds the struts. The concentration is highest close to the struts and is relatively low close to the lumen between the stent struts. The concentration field around the first strut is asymmetric and skewed upstream. This is due to the fact that the endothelium is denuded between the stent struts but intact upstream of the stent. Crossing the intact endothelium, the pressure drops by more than 50 % from the lumen to the arterial wall, while in between struts, the pressure drop is virtually negligible. Thus, convection is higher in between struts than at the upstream and downstream ends of the stent, creating a backflow pattern around the strut that distorts the symmetry in the concentration distribution.
Over the course of the next few days, the concentration distribution pattern homogenizes in the media while the SES rapidly empties of drug. After 1 day, the concentration level in the media has already dropped by an order of magnitude compared to the maximum concentration. This is due to the fact that the fast-release kinetics of the baseline stent has already emptied the entire drug supply at this point. After a week, the maximum concentration in the therapeutic domain has dropped by two orders of magnitude. The radial concentration distribution is now skewed towards the adventitia (rather than the intima) while a small concentration reservoir has formed upstream of the stent where convective transport forces are not as strong because the endothelium is not denuded.
Figure 2b depicts the temporal evolution of the spatially averaged NMC in the SES and media. Peak concentration in the SES occurs minutes after stent implantation and attains only a quarter of the peak concentration in the media. Within the first day, the drug vanishes from the SES. The high concentration in the media (\(\approx \)8 \(\,\times \,10^{-3}\)) is followed by a rapid drop within the following \(12\,\hbox {h}\) to an NMC of \(1\,\times \,10^{-3}\), which corresponds well to the value of the maximum binding site density of paclitaxel (\(b_\mathrm{max}=0.127\)). The decrease then slows down considerably for the remainder of the week.
3.2 Sensitivity analysis of the model
In light of the uncertainty in some of the transport model parameters, we studied the sensitivity of the baseline results to the drug diffusivity coefficients in the SES and the media while maintaining the ratio of the Damköhler number and the Peclet number constant. Given that all equations are non-dimensional, the analysis is presented in terms of the sensitivity of the NMC to the Peclet number in the different layers:
Figure 3 illustrates the sensitivity of the model results to the Peclet number in the SES as well as to the radial and axial Peclet numbers in the media. We have selected two parameters to measure the sensitivity of the model: (1) the magnitude of the peak NMC in the SES and in the media (Fig. 3a, c, e) and (2) the ratio of the NMC after 1, 5, and 7 days compared to the maximum value (Fig. 3b, d, f); this retention coefficient quantifies the drop following maximum concentration in the media.

Sensitivity analysis (paclitaxel). Dependence of maximum NMC in the SES and media on A SES Peclet number, C radial medial Peclet number, and E axial medial Peclet number. Dependence of retention coefficient at three time points (\(\tilde{t}=1\,\hbox {day}, \tilde{t}=5\,\hbox {day}\), and \(\tilde{t}=7\,\hbox {day}\) post-stenting) on B SES Peclet number, D radial medial Peclet number, and F axial medial Peclet number
Varying the Peclet number in the SES over several orders of magnitude has a fairly small effect on the concentration levels in the SES and media (Fig. 3a). More specifically, varying the SES Peclet number over four orders of magnitude leads to an increase in the maximum NMC of \(\approx \)55 % in the SES and only \(\approx \)10 % in the media. The retention coefficient at 1 day changes only by 6 % (Fig. 3b). The NMC in the wall is more sensitive to a variation of the radial Peclet number in the media (Fig. 3c, d). While the peak NMC in the SES changes by only a moderate 32 %, the peak concentration in the media changes by \(\approx \)300 %. The highest sensitivity is observed as one goes from \( Pe ^r_{\mathrm{m}}=0.02\) to \( Pe ^r_{\mathrm{m}}=0.2\). With the peak concentration increasing, the retention coefficient at 1 day decreases by 75 % over the entire \( Pe ^r_{\mathrm{m}}\) range. Here, too, the majority of the variation occurs in the \( Pe ^r_{\mathrm{m}}<1\) domain. The retention coefficients at 5 and 7 days decrease by more moderate values of \(\approx \)45 and \(\approx \)34 %, respectively.
The NMC in the SES appears to be largely insensitive to the axial Peclet number in the media (Fig. 3e), whereas the medial NMC is weakly affected, with a \(\approx \)14 % total decrease over a two-order-of-magnitude change in \( Pe ^z_{\mathrm{m}}\). The retention coefficients for all three time points decrease by \(\approx \)30 %. Interestingly, in all cases the retention coefficients at 5 and 7 days (Fig. 3b, d, f) are less sensitive to Peclet number variations than the day one retention values, implying that the long-term evolution is affected differently than the short term.
A value commonly used in computational models for the medial porosity is \(\varepsilon _\mathrm{m}=0.25\). However, the porosity of the media largely depends on its state of health. With the fiber matrix porosity of the media fixed at \(\varepsilon _\mathrm{f,m}=0.45\), we varied \(\varepsilon _\mathrm{m}\) in the range of 0.1–0.4 so that the model remains consistent with its definition of the volume fraction of SMCs \(\chi _\mathrm{SMC}\). Figure 4 depicts the sensitivity of the peak concentrations in the SES and the media (Fig. 4a) and the retention coefficient in the media (Fig. 4b) to \(\varepsilon _\mathrm{m}\). The SES concentration is unaffected by the medial porosity; however, the maximum NMC in the media more than doubles in the considered \(\varepsilon _\mathrm{m}\) interval. Accordingly, the magnitude of the drop at one day increases by 38 %. Drug retention at \(5\) and 7 days is largely unaffected by the porosity variations.

Sensitivity analysis (paclitaxel). A Dependence of maximum normalized mean concentration (NMC) in the subendothelial space (SES) and media on medial porosity. B Dependence of the retention coefficient at three time points: \(\tilde{t}=1\,\hbox {day}\), \(\tilde{t}=2\,\hbox {days}\), and \(\tilde{t}=7\,\hbox {days}\) post-stenting on medial porosity
Changing the lumenal Reynolds number in the physiological range (\( Re _\mathrm{l} \in \left[ 100,800\right] \)) reveals a significant dependence of the concentration evolution in the SES on blood flow in the lumen (Fig. 5). The maximum NMC in the SES decreases significantly (more than 85 %) with increasing Reynolds number; however, consistent with the previous results by Mongrain et al. (2007), the maximum total NMC in the media is only weakly affected.

Sensitivity analysis (paclitaxel). Dependence of the maximum NMC in the SES and media on Reynolds number in the lumen
3.3 Effect of one-layer modeling
It is currently common in DES modeling to treat the arterial wall as a single layer with homogenized transport properties mostly representing the media. However, it was shown by Hwang and Edelman (2002) that the layered structure of the arterial wall can affect the deposition and distribution of drugs. Figure 6 illustrates the differences in the concentration distribution occurring when a one-layer model (OLM) is used instead of the two-layer model (TLM).

One-layer versus two-layer modeling (paclitaxel). Normalized velocity (NV) and normalized concentration (NC) field around the first stent strut for a situation where the SES has been thickened by a factor of 3 and the total stent strut size has been decreased to \(150\,\upmu \hbox {m}\). Time shown is the point of maximum concentration in the media
To emphasize the differences in the predictions of the flow field (and consequently the concentration field) resulting from the TLM and OLM, we altered the geometric setup of our models: In both models we reduced the total strut size from the baseline diameter of \(d_\mathrm{s}=\text {metallic strut diameter}+2\cdot \text {polymer thickness}=0.15\,\hbox {mm}+2\times 0.05\,\hbox {mm}=0.25\,\hbox {mm}\) to a diameter of \(d_\mathrm{s}=0.05\,\hbox {mm}+2\times 0.025\,\hbox {mm}=0.15\,\hbox {mm}\) (representing a second-generation DES strut) and considered a case where the SES is thickened by a factor of three compared to the baseline situation due to the development of atherosclerosis. Figure 6 shows the superimposed flow and drug concentration field around the first strut of the stent. Both the TLM and OLM predict a back-flow pattern around the first strut; however, the TLM predicts a more pronounced back flow whereby the back-flowing fluid enters the SES, while the OLM is incapable of capturing this characteristic flow feature. We can also see that the local changes in the flow field affect the concentration distribution and magnitude close to the stent strut.
Figure 7 compares the OLM and TLM predictions for the NMC in the SES and in the media for the baseline model.

One-layer versus two-layer modeling (paclitaxel). NMC for two different SES thicknesses (\(L_\mathrm{ses}=L_\mathrm{ses,0}\) and \(L_\mathrm{ses}=3 L_\mathrm{ses,0}\)). A In the SES (both predictions of the OLM collapse onto one curve) B in the media
Using Eq. (25) recovers the qualitative temporal evolution of the NMC in the SES. However, the peak concentration is underestimated by about 30 %, and this offset continues to grow to \(\approx \)100 % at 2 h post-implantation. For a “diseased” configuration with a threefold thickened SES, the differences between the TLM and OLM become yet more pronounced, and the peak concentration is underestimated by 100 %. In the media, the agreement is very good qualitatively and quantitatively. Here also the discrepancies grow when the SES thickness is increased.
3.4 Effect of reaction modeling
We wished to establish whether an equilibrium reaction model adequately represents drug transport in the arterial wall. Figure 8 compares the temporal evolution of the total concentration (NMC) in the media for the reversible binding reaction model to the case where free and bound drug are assumed to be in a constant equilibrium. Using an equilibrium assumption, the very early behavior predicted by the dynamic model can be qualitatively recovered. However, the peak concentration predicted by the equilibrium model occurs considerably later and is significantly higher compared to the dynamic reversible binding model. For the commonly used value of \(\kappa =20\) (Vairo et al. 2010), the peak concentration is \(\approx \)3 times higher than that predicted by the dynamic reversible reaction model and occurs \(\approx \)9 h later. With increasing \(\kappa \), the peak becomes progressively higher and is predicted to occur later. In all cases including the dynamic reaction model, the arterial wall becomes completely void of drug within a week of stent implantation. The emptying time is quicker for smaller values of \(\kappa \).

Temporal evolution of the total NMC in the media as predicted by a reversible binding reaction model (baseline model) compared to an equilibrium model for different values of the total binding coefficient \(\kappa \)
3.5 Effect of the choice of drug
All results presented thus far focused on paclitaxel. Another common drug used in DES is sirolimus. Although different in their mode of action in preventing smooth muscle cell (SMC) proliferation and migration (Garg and Serruys 2010), sirolimus and paclitaxel have similar transport properties (see Table 3). With a binding potential \(B_\mathrm{p}>40\), both drugs fall in the category of strongly retained drugs (Tzafriri et al. 2009).
Figure 9 compares the distribution of the bound drug fraction (\(b\)) in the upstream region of the therapeutic domain for paclitaxel and sirolimus at the time of maximum total NMC in the media (\(\tilde{t}=50\,\hbox {min}\)) and 1 day after stent implantation.

Paclitaxel versus sirolimus. Bound drug fraction \(b\) at two different time points: \(\tilde{t}=50\,\hbox {min}\) (first row) and \(\tilde{t}=1\,\hbox {day}\) (second row)
Within the first hour, binding has occurred over the entire width of the arterial wall for both drugs. Binding is maximal close to the stent struts and drops gradually with distance away from the polymer. The concentration gradient is sharper in the case of sirolimus. For paclitaxel, there is a region near the lumenal surface in between the struts where little drug binding occurs; such a region exists but is considerably smaller in the case of sirolimus. At 1 day, the binding pattern has spread further upstream. Interestingly, paclitaxel has invaded a larger portion of the upstream therapeutic domain than sirolimus, although the axial diffusion coefficient of sirolimus is larger. These observations underscore the more convection-dominated transport of paclitaxel compared with sirolimus. Following the emptying of the stent, the unbinding process becomes the dominant effect: The binding fraction in the case of paclitaxel drops to \(\approx \)70 % on average, while the equivalent value for sirolimus is \(\approx \)60 %.
Figure 10 compares drug transport characteristics for stents loaded with either paclitaxel or sirolimus for the baseline fast-release case (panels A, C, and E) and for the case of a slow-release stent (panels B, D and F).

Paclitaxel versus sirolimus. Temporal evolution of the NMC in the SES for a, A fast-release and B slow-release stent. NMC in the media for a, C fast-release and D slow-release stent. Integral Damköhler number for a, E fast-release and F slow-release stent (the horizontal line indicates the \( Da _\mathrm{int}=1\) threshold)
Figure 10a, c demonstrates that for the fast-release stent, the differences between paclitaxel and sirolimus are rather minor. Peak concentration magnitude and timing in the SES and media are comparable. The one difference is the slower drop following peak concentration in the media in the case of sirolimus compared with paclitaxel: For paclitaxel, the drop to 25 % of the peak average concentration takes \(\approx \)6 h, while for sirolimus, the same drop almost requires 1 day.
The slow-release stent is geometrically identical to the fast-release stent; only the diffusion coefficient in the polymer coating has been reduced by three orders of magnitude to \(\tilde{D}_\mathrm{c}=1\times 10^{-16}\,\hbox {m}^{2}\,\hbox {s}^{-1}\). When comparing the concentration profiles for the two drugs in the SES (Fig. 10b) and media (Fig. 10d) for the slow-release stent, a very different picture emerges: While the qualitative behavior for both drugs remains similar, the concentration levels of sirolimus are continuously above those of paclitaxel. At peak concentration in the SES, the NMC of sirolimus is \(\approx \)50 % higher than that of paclitaxel. This discrepancy grows to more than 100 % at 8 weeks. The peak concentration of sirolimus in the media is three times the peak concentration of paclitaxel, and after 8 weeks, the NMC remains more than twice as high.
Figure 10e, f compares the time evolution of the integral Damköhler number for both drugs for the fast-release and slow-release stent platforms. In the fast-release case, paclitaxel begins in the convection-dominated regime (\( Da _\mathrm{int}<1\)) and after the first half-day transitions into a slightly more binding-dominated regime. The transition is faster for sirolimus since with the exception of the first few hours after the beginning of drug elution, the transport of sirolimus is dominated by the binding and unbinding process. For the slow-release stent, the balance of convective and reactive terms is less variable: Paclitaxel remains in a regime where convection and binding/unbinding are of comparable importance, whereas sirolimus is clearly dominated by the binding/unbinding process over the entire time.
4 Discussion
4.1 Drug release is coupled to the drug and its transport properties in the arterial wall
Considering the differences in the binding and unbinding timescales of paclitaxel and sirolimus (see Table 5), it might appear surprising that both drugs behave so similarly in the case of a fast-release stent platform. The integral Damköhler number sheds light on this: For paclitaxel, the drug is initially predominantly in the free (and thus mobile) state and invades the media driven by plasma flow in the arterial wall, since the timescale of convection is faster than that for binding. This phase determines the initial distribution of the drug within the arterial wall. With rising concentration levels of free drug in the media, the binding rate increases, leading to an increased \( Da _\mathrm{int}\) and a transitional passage through a phase of competition between the binding and convection terms. Once all binding sites are saturated, any excess drug is washed out. As soon as the stent polymer is empty and the arterial wall has become void of all excess drugs, the transport becomes dominated by unbinding since \(\tilde{t}_\mathrm{rR}\gg \tilde{t}_\mathrm{C}\) and \(\tilde{t}_\mathrm{rR}\gg \tilde{t}_\mathrm{D}\). For sirolimus, on the other hand, the initial dominant process is binding. When the drug enters the media, binding sites are very rapidly occupied, since \(\tilde{t}_\mathrm{fR}\) is several orders of magnitude smaller than \(\tilde{t}_\mathrm{C}\) or \(\tilde{t}_\mathrm{D}\), and only drug exceeding the maximum binding capacity invades new areas of the media. At the same time, this exhaustion of binding sites reduces the binding rate, which initiates a shorter, transient passage through a more convection-driven phase. Once the stent polymer coating is empty, the behavior becomes dominated by the unbinding of drug from its binding sites, since the timescale of the unbinding process is longer than that of either convection or diffusion.
As a result, we can subdivide the activity of DES releasing hydrophobic drugs into two phases: an initial release phase where new drug is supplied from the stent and a secondary post-release phase where the drug effect is predominantly determined by the binding and unbinding process. The first phase is prone to changes in the convective and diffusive transport of the free drug. The relevance of this sensitivity is determined by the Damköhler number and the Peclet number and depends not only on the release rate (determined by \(\tilde{D}_\mathrm{c}\)) but also on the initial concentration in the polymer \(\tilde{c}_0\). As demonstrated by the evolution of the integral Damköhler number for the fast-release stent, even the highly reactive sirolimus transport can become convection-driven, which explains the similarity in concentration distributions of both drugs. The sensitivity analysis of the radial Peclet number in the media \( Pe ^r_{\mathrm{m}}\) and the medial porosity \(\varepsilon _\mathrm{m}\) illustrates this nicely: In the first case, we do not simply vary the relative importance of diffusion and convection but also bring the Damköhler number down from a regime where binding/unbinding dominates to a diffusion-dominated regime, and thus, we get such a large variability for the low \( Pe ^r_{\mathrm{m}}\) range. Once \( Da >1\), the results become almost insensitive to further variation. The same is true for the variation of the medial porosity where effectively the ratio of the Damköhler and Peclet number is varied: Going from a low to a high porosity, we approach \({ Da }/{ Pe ^r_{\mathrm{m}}}\approx 1\), and the peak variability goes down. At the same time, the retention coefficient almost does not vary since, with the free drug gone, it is solely determined by the unbinding process.
Once we reduce the drug release rate significantly, we obtain a very different picture: The slow, long-lasting drug supply balances the otherwise rapid unbinding process of sirolimus, ensuring a high, only slowly decaying concentration level. The transport of paclitaxel does not benefit as significantly from the more permanent release since the very slow timescale of unbinding determines the retention characteristics independent of the release kinetics.
The present results demonstrate that paclitaxel and sirolimus have widely different transport dynamics in the arterial wall. These differences suggest that different drug delivery strategies should be used for these drugs. Moreover, both drugs have different therapeutic behavior: Paclitaxel has a broad therapeutic window with a minimum effective concentration of \(\tilde{c}_\mathrm{eff}=1\times 10^{-5}\,\hbox {mol}\,\hbox {m}^{-3}\) and a maximum concentration level of \(\tilde{c}_\mathrm{tox}=1\times 10^{-2}\,\hbox {mol}\,\hbox {m}^{-3}\) above which the drug becomes toxic (McGinty et al. 2011; Kamath et al. 2006), while for sirolimus to be effective, saturation of the FKBP12 binding sites appears to be required (Wang et al. 2007), raising the minimum effective concentration significantly. Thus, the choice of a drug delivery strategy for a particular drug becomes a crucial design parameter in DES development. Based on the present results and for the present geometric and flow conditions considered, a paclitaxel stent platform should optimally employ fast-release kinetics with a low initial drug load, whereas a sirolimus stent should target slow-release kinetics with a higher initial drug load. It was well established by Balakrishnan et al. (2007) that drug release kinetics are an important factor in the DES design process. Our results complement those findings and highlight the close coupling between the drug chosen and the applied drug delivery strategy.
The interplay between stent, flow, and drug parameters provides an opportunity for implementing sophisticated optimization strategies for targeting “desirable” drug concentration profiles in the arterial wall. Optimal drug profiles would be defined in terms of various criteria including how uniformly the drug is distributed within the arterial wall and whether or not drug concentrations fall within the therapeutically efficacious window.
An additional conclusion from the present results is that the uncertainty in the diffusion coefficients in the arterial wall is of a relatively minor importance for the concentration distribution as long as the relevant dimensionless quantities, most notably the Peclet and Damköhler numbers, remain in the appropriate physiological range.
4.2 Multilayer model offers improved accuracy for pathological situations
Delayed re-endothelialization occurring with DES motivates the need for more detailed drug concentration information, especially close to the endothelial surface. The SES and media are characterized by fundamentally different transport properties (Table 3). Accounting for these differences in the TLM revealed that higher concentration levels can be obtained in the SES than in the media especially early after stent implantation (Fig. 7a). The strong sensitivity of the SES drug concentration to the lumenal flow field (Fig. 5) underscores the differences between the two layers of the arterial wall. This also indicates that assuming a steady arterial flow may not be sufficient to make accurate predictions for the concentration levels in the arterial wall close to the lumen. In the future, time-dependent flow computations should investigate this issue.
A detailed description of drug concentration within the SES is only available in the TLM. The concentration in the SES as an averaged quantity can be extracted from the OLM with the presented approach (Eq. 25). Except for an offset, the qualitative behavior of this global metric is very well recovered and even the distribution pattern at the endothelial surface is in good agreement between the TLM and OLM cases. The reason for this offset is the absence of the drug flux from the stent polymer entering the SES in the model equation. Accordingly, the offset increases with thickening of the SES where the drug flux from the polymer into the SES increases due to the larger contact area between the SES and polymer. This weakness could be overcome by adding an averaged source term to the equation (Eq. 31). However, averaged concentration levels might not be the only information necessary for unraveling the processes leading to delayed re-endothelialization, especially when incomplete endothelial coverage can be observed even up to five years after DES implantation (Räber et al. 2011). As the diseased configuration in Fig. 6a demonstrated, predictions of the flow field diverge significantly depending on the model. Convective forces can contribute considerably to the drug distribution in the arterial wall when free drug levels are elevated (Figs. 9, 10e).
The representation of a diseased arterial wall as presented in this study is drastically simplified for demonstration purposes. In its early stages, atherosclerosis affects primarily the intima and as such the TLM offers the potential to account for different diseased states. For example, the presence of SMCs that have migrated into the SES can be modeled by adding a reaction term to the transport equation in the SES with customized reaction parameters (like the maximum binding site concentration) or changes to the transport parameters in the SES. This could also enable improved evaluation of experimental results that are often performed on healthy vessels (Radeleff et al. 2010) and facilitate conjectures to the diseased case.
4.3 Reduction in the reaction model fails to capture important features of the transport dynamics
As we could see, the drug transport process is highly dynamic and intertwined with the release process. From a computational point of view, it would certainly be desirable, if possible, to avoid the stiff (especially in the case of sirolimus) and computationally expensive reaction equation. Tzafriri et al. (2009) explored several possible concentration-dependent simplifications of the dynamic reaction model. As considered here, the crudest simplification to account for drug binding is to assume a constant partitioning of bound and free drug inherent in the equilibrium model (Eq. 9). Figure 8 illustrates that an equilibrium reaction model fails to capture essential features of the transport process: The binding coefficient \(\kappa \) reduces the transport term permanently, and thus, the predicted accumulation of drug far exceeds that of the dynamic reaction model. Moreover, the coupling of the convection–diffusion and reaction equations transfers free (i.e., mobile) drug into a bound (i.e., stored) state. This cannot be reflected with the equation obtained by the equilibrium model, and thus, the residence time of the drug in the arterial wall is underestimated. Matching the prediction of the drug accumulation by the equilibrium model to that of the dynamic reaction model requires a reduction in the partition coefficient, while matching the drug residence time demands an increase. Both objectives cannot be achieved simultaneously.
Abbreviations
- \(b\) :
-
Superficially averaged bound concentration
- \(b_\mathrm{max}\) :
-
Maximum binding site density
- \(B_\mathrm{p}\) :
-
Binding potential
- \(c_0\) :
-
Initial concentration in the stent polymer
- \(c_j\) :
-
Superficially averaged free concentration in layer \(j\)
- \(c_\mathrm{T}\) :
-
Superficially averaged total concentration
- \(d_\mathrm{s}\) :
-
Strut diameter
- \( Da \) :
-
Damköhler number in the media
- \(D_j^i\) :
-
Effective diffusion coefficient in layer \(j\) in direction \(i\) (isotropic diffusion assumed when no superscript)
- \(f_\mathrm{cb}\) :
-
Ratio of initial concentration and maximum binding site density
- \(J_{\mathrm{s,}j}\) :
-
Solute flux through membrane \(j\)
- \(J_{\mathrm{v,}j}\) :
-
Fluid flux through membrane \(j\)
- \(k_\mathrm{f}\) :
-
Drug binding rate coefficient
- \(k_\mathrm{p}\) :
-
Concentration-dependent partition coefficient
- \(k_\mathrm{r}\) :
-
Drug unbinding rate constant
- \(k^*_\mathrm{r}\) :
-
Drug unbinding rate constant corrected for solid tissue fraction
- \(K_\mathrm{d}\) :
-
Equilibrium dissociation constant
- \(L_j\) :
-
Thickness of layer \(j\)
- \(L_{\mathrm{p,}j}\) :
-
Hydraulic conductivity of membrane \(j\)
- \(M_i\) :
-
Molecular weight of drug \(i\)
- \(p_j\) :
-
Pressure in layer \(j\)
- \(P_j\) :
-
Permeability of layer \(j\)
- \(P_{\mathrm{D},j}\) :
-
Darcy permeability in layer \(j\)
- \( Pe _j^i\) :
-
Peclet number in layer \(j\) in direction \(i\) (isotropic Peclet number assumed when no superscript)
- \( Pe _j^*\) :
-
Membrane Peclet number in membrane \(j\)
- \(r,z\) :
-
Radial and axial coordinates
- \(r_\mathrm{f}\) :
-
Average radius of fibers of the extracellular matrix
- \(r_\mathrm{mol}^i\) :
-
Average molecular radius of drug \(i\)
- \( Re \) :
-
Reynolds number
- \( Re _\mathrm{l}\) :
-
Lumenal Reynolds number
- \( Re _\mathrm{d}\) :
-
Reynolds number based on the hydrodynamic strut diameter
- \( Sc \) :
-
Schmidt number
- \(s_j\) :
-
Sieving coefficient in membrane \(j\)
- \(t\) :
-
Time
- \(t_\mathrm{E}\) :
-
Emptying/release time
- \(\varvec{u}=\left( u\;v\;w \right) ^T\) :
-
Velocity
- \(V_j\) :
-
Volume of layer \(j\)
- \(\gamma _j\) :
-
Hindrance coefficient in layer \(j\)
- \(\varGamma _j\) :
-
Boundary \(j\)
- \(\delta _\mathrm{c}\) :
-
Concentration boundary layer thickness
- \(\varepsilon _j\) :
-
(Effective) porosity of layer \(j\)
- \(\varepsilon _{\mathrm{f,}j}\) :
-
Fiber matrix porosity of layer \(j\)
- \(\kappa \) :
-
Total binding coefficient
- \(\varLambda _j\) :
-
Lag coefficient in layer \(j\)
- \(\mu \) :
-
Fluid dynamic viscosity
- \(\rho \) :
-
Fluid density
- \(\sigma _{\mathrm{f,}j}\) :
-
Staverman reflection coefficient in membrane \(j\)
- \(\varPhi _{\mathrm{f,}j}\) :
-
Reduction coefficient
- \(\chi _\mathrm{SMC}\) :
-
Volume fraction of smooth muscle cells
- 0:
-
Reference value
- b:
-
Blood
- e:
-
Endothelium
- eel:
-
External elastic lamina
- iel:
-
Internal elastic lamina
- l:
-
Lumen
- m:
-
Media
- p:
-
Plasma
- PAX:
-
Paclitaxel
- ses:
-
Subendothelial space
- SIR:
-
Sirolimus
- SMC:
-
Smooth muscle cells
- th:
-
Therapeutic domain
- w:
-
Arterial wall
- \(\alpha \) :
-
Liquid phase
- BDF:
-
Backward differentiation formula
- BLE:
-
Boundary layer elements
- BMS:
-
Bare-metal stents
- DCB:
-
Drug-coated balloons
- DES:
-
Drug-eluting stents
- EEL:
-
External elastic lamina
- FEM:
-
Finite element method
- IEL:
-
Internal elastic lamina
- NC:
-
Normalized concentration
- NMC:
-
Normalized mean concentration
- NV:
-
Normalized velocity
- OLM:
-
One-layer model
- RHS:
-
Right-hand side
- SCAAR:
-
Swedish Coronary Angiography and Angioplasty Registry
- SES:
-
Subendothelial space
- SMC:
-
Smooth muscle cell
- TLM:
-
Two-layer model
References
Ai L, Vafai K (2006) A coupling model for macromolecule transport in a stenosed arterial wall. Int J Heat Mass Transf 49(9–10):1568–1591
AIAA (1998) AIAA guide for the verification and validation of computational fluid dynamics simulations. American Institute of Aeronautics and Astronautics
Balakrishnan B, Tzafriri AR, Seifert P, Groothuis A, Rogers C, Edelman ER (2005) Strut position, blood flow, and drug deposition—implications for single and overlapping drug-eluting stents. Circulation 111(22):2958–2965
Balakrishnan B, Dooley JF, Kopia G, Edelman ER (2007) Intravascular drug release kinetics dictate arterial drug deposition, retention, and distribution. J. Control. Release 123(2):100–108
Borghi A, Foa E, Balossino R, Migliavacca F, Dubini G (2008) Modelling drug elution from stents: effects of reversible binding in the vascular wall and degradable polymeric matrix. Comput Methods Biomech Biomed Eng 11(4):367–377
Curry FRE (1984) Mechanics and thermodynamics of transcapillary exchange. In: Renkin E, Michel C (eds) The cardiovascular system. Microcirculation, handbook of physiology, vol 1. American Physiological Society, Bethesda, MD, pp 309–374
Feenstra PH, Taylor CA (2009) Drug transport in artery walls: a sequential porohyperelastic-transport approach. Comput Methods Biomech Biomed Eng 12(3):263–276
Formaggia L, Quarteroni A, Veneziani A (eds) (2009) Cardiovascular mathematics. Springer Milan, Milano
Garg S, Serruys PW (2010) Coronary stents: current status. J Am Coll Cardiol 56(10, Supplment 1):S1–S42
Gray WA, Granada JF (2010) Drug-coated balloons for the prevention of vascular restenosis. Circulation 121(24):2672–2680
Hara H, Nakamura M, Palmaz JC, Schwartz RS (2006) Role of stent design and coatings on restenosis and thrombosis. Adv Drug Deliv Rev 58(3):377–386
Hilder T, Hill J (2008) Probability of encapsulation of paclitaxel and doxorubicin into carbon nanotubes. Micro Nano Lett 3(2):41
Hindmarsh AC, Brown PN (2005) SUNDIALS: suite of nonlinear and differential/algebraic equation solvers
Huang Y, Rumschitzki D, Chien S, Weinbaum S (1994) A fiber matrix model for the growth of macromolecular leakage spots in the arterial intima. J Biomech Eng 116(4):430–445
Hwang CW, Edelman ER (2002) Arterial ultrastructure influences transport of locally delivered drugs. Circ Res 90(7):826–832
Hwang CW, Wu D, Edelman ER (2001) Physiological transport forces govern drug distribution for stent-based delivery. Circulation 104(5):600–605
Kamath KR, Barry JJ, Miller KM (2006) The Taxus(TM) drug-eluting stent: a new paradigm in controlled drug delivery. Adv Drug Deliv Rev 58(3):412–436
Karner G, Perktold K, Zehentner HP (2001) Computational modeling of macromolecule transport in the arterial wall. Comput Methods Biomech Biomed Eng 4(6):491–504
Kedem O, Katchalsky A (1958) Thermodynamic analysis of the permeability of biological membranes to non-electrolytes. Biochim Biophys Acta 27:229–246
Khakpour M, Vafai K (2008) Critical assessment of arterial transport models. Int J Heat Mass Transf 51(3–4):807–822
LaDisa JF (2004) Stent design properties and deployment ratio influence indexes of wall shear stress: a three-dimensional computational fluid dynamics investigation within a normal artery. J Appl Physiol 97(1):424–430
Levin AD, Vukmirovic N, Hwang CW, Edelman ER (2004) Specific binding to intracellular proteins determines arterial transport properties for rapamycin and paclitaxel. Proc Natl Acad Sci USA 101(25):9463–9467
Levitt DG (1975) General continuum analysis of transport through pores: I. Proof of Onsager’s reciprocity postulate for uniform pore. Biophys J 15(6):533–551
Lovich MA, Creel C, Hong K, Hwang CW, Edelman ER (2001) Carrier proteins determine local pharmacokinetics and arterial distribution of paclitaxel. J Pharm Sci 90(9):1324–1335
McGinty S, McKee S, Wadsworth RM, McCormick C (2011) Modelling drug-eluting stents. Math Med Biol 28(1):1–29
Mongrain R, Brunette J, Faik I, Bulman-Feleming N, Nguyen T (2005) Numerical modeling of coronary drug eluting stents. In: Suri J (ed) Plaque imaging: pixel to molecular level. IOS Press, Amsterdam, Washington, DC, pp 443–458
Mongrain R, Faik I, Leask RL, Rodes-Cabau J, Larose E, Bertrand OF (2007) Effects of diffusion coefficients and struts apposition using numerical simulations for drug eluting coronary stents. J Biomech Eng 129(5):733–742
O’Connell MK, Murthy S, Phan S, Xu C, Buchanan J, Spilker R, Dalman RL, Zarins CK, Denk W, Taylor CA (2008) The three-dimensional micro- and nanostructure of the aortic medial lamellar unit measured using 3D confocal and electron microscopy imaging. Matrix Biol 27(3):171–181
Pontrelli G, de Monte F (2010) A multi-layer porous wall model for coronary drug-eluting stents. Int J Heat Mass Transf 53(19–20):3629–3637
Prosi M, Zunino P, Perktold K, Quarteroni A (2005) Mathematical and numerical models for transfer of low-density lipoproteins through the arterial walls: a new methodology for the model set up with applications to the study of disturbed lumenal flow. J Biomech 38(4):903–917
Radeleff B, Lopez-Benitez R, Stampfl U, Stampfl S, Sommer C, Thierjung H, Berger I, Kauffmann G, Richter GM (2010) Paclitaxel-induced arterial wall toxicity and inflammation: tissue uptake in various dose densities in a minipig model. J Vasc Interv Radiol 21(8):1262–1270
Räber L, Wohlwend L, Wigger M, Togni M, Wandel S, Wenaweser P, Cook S, Moschovitis A, Vogel R, Kalesan B, Seiler C, Eberli F, Lüscher TF, Meier B, Jüni P, Windecker S (2011) Five-year clinical and angiographic outcomes of a randomized comparison of sirolimus-eluting and paclitaxel-eluting stents/clinical perspective. Circulation 123(24):2819–2828
Sarno G, Lagerqvist B, Fröbert O, Nilsson J, Olivecrona G, Omerovic E, Saleh N, Venetzanos D, James S (2012) Lower risk of stent thrombosis and restenosis with unrestricted use of ’new-generation’ drug-eluting stents: a report from the nationwide Swedish coronary angiography and angioplasty registry (SCAAR). Eur Heart J 33(5):606–613
Seo T, Schachter LG, Barakat AI (2005) Computational study of fluid mechanical disturbance induced by endovascular stents. Ann Biomed Eng 33(4):444–456
Tzafriri AR, Levin AD, Edelman ER (2009) Diffusion-limited binding explains binary dose response for local arterial and tumour drug delivery. Cell Prolif 42(3):348–363
Tzafriri AR, Groothuis A, Price GS, Edelman ER (2012) Stent elution rate determines drug deposition and receptor-mediated effects. J Control Release 161(3):918–926
Vairo G, Cioffi M, Cottone R, Dubini G, Migliavacca F (2010) Drug release from coronary eluting stents: a multidomain approach. J Biomech 43(8):1580–1589
Vergara C, Zunino P (2008) Multiscale boundary conditions for drug release from cardiovascular stents. Multiscale Model Simul 7(2):565–588
Wang XT, Venkatraman S, Boey F, Loo SC, Tan LP (2007) Effects of controlled-released sirolimus from polymer matrices on human coronary artery smooth muscle cells. J Biomater Sci Polym Ed 18(11):1401–1414
Weinbaum S, Tzeghai G, Ganatos P, Pfeffer R, Chien S (1985) Effect of cell turnover and leaky junctions on arterial macromolecular transport. Am J Physiol Heart Circ Physiol 248(6):H945–H960
Whitaker S (1986) Transient diffusion, adsorption and reaction in porous catalysts: the reaction controlled, quasi-steady catalytic surface. Chem Eng Sci 41(12):3015–3022
Whitaker S (2010) The method of volume averaging. Kluwer Academic Publishers, Dordrecht
Yang N, Vafai K (2006) Modeling of low-density lipoprotein (LDL) transport in the artery-effects of hypertension. Int J Heat Mass Transf 49(5–6):850–867
Zunino P (2004) Multidimensional pharmacokinetic models applied to the design of drug-eluting stents. Cardiovasc Eng 4(2):181–191
Acknowledgments
Franz Bozsak is supported by a doctoral fellowship from Ecole Polytechnique. This work is supported in part by a permanent endowment in Cardiovascular Cellular Engineering from the AXA Research Fund.
Author information
Authors and Affiliations
Corresponding author
Appendices
Appendix 1: From the Brinkman equation to Darcy’s law
Fluid flow in the porous layers of the arterial wall can be described by the standard Darcy–Brinkman equation
An order-of-magnitude analysis on the Brinkman equation reveals that the viscous term only plays a significant role when \(\tilde{L}^2 \sim \tilde{P}_\mathrm{D}\). Thus, the effect of the viscous term is limited to a boundary layer which is an order of magnitude thinner than the endothelium. Consequently, for both layers (the subendothelial space and the media) the governing equation is simplified to Darcy’s original formulation:
Appendix 2: Derivation of the weighted concentration
The local average fluid concentration \(\bar{c}_{\alpha ,j}\) as required by the Kedem–Katchalsky matching boundary conditions is calculated from the integral of the solution of the attributed differential equation for the concentration distribution in one dimension (Levitt 1975):
with
Given strictly radial transport from domain \(j-1\) to \(j+1\), this expression allows an approximate evaluation of the average concentration in the respective “layer” \(j\). When the exponential function is expanded to third order, it becomes obvious that this expression reflects the weighted average concentration of the layer:
Appendix 3: Including reaction at the endothelial surface
Like smooth muscle cells, endothelial cells interact with the drugs eluted from the stent surface. Thus, we wish to include this interaction in the boundary condition at the endothelium via a concentration-dependent partition coefficient. The Kedem–Katchalsky equation is derived assuming steady-state conditions (Levitt 1975). Assuming \(\frac{\partial b}{\partial t}=0\) for the reaction equation (Eq. 10) yields
Reintroducing this expression into the transport equation of the arterial wall (Eq. 7) leads to the concentration-dependent partition coefficient:
Assuming an instantaneous reaction of the drug on the surface of ECs, the intrinsic fluid concentration used in the Kedem–Katchalsky equation is now:
Appendix 4: Governing equation for the equilibrium case
Assuming constant equilibrium between bound and free drug concentration (Eq. 9), the governing equation (Eq. (7)) reduces to:
where \(c_{\mathrm{T,}j}\) represents the total drug concentration, the sum of the free concentration \(c_j\) and the bound concentration \(b\): \(c_{\mathrm{T,}j}=b+c_j=\kappa c_j\).
Appendix 5: Definition of porosities
The (effective) porosity \(\varepsilon _j\) of layer \(j\) describes the ratio of the pore space volume to the total volume:
The fiber matrix porosity \(\varepsilon _{\mathrm{f,}j}\) of layer \(j\) describes the following volume ratios:
which for the media needs to be adjusted by the volume fraction of SMCs
to yield the effective porosity in the media
In the subendothelial space, the (effective) porosity equals the fiber matrix porosity due to the absence of SMCs.
Appendix 6: Remaining drug-dependent parameters of the model
Except for the lag coefficient \(\varLambda _j\), we followed in our derivation of the remaining model parameters the description of Formaggia et al. (2009). The lag coefficient \(\varLambda _j\) of the convective transport term which results from averaging can be calculated as:
where \(\gamma _j \le 1\) is the hindrance coefficient (in layer \(j\)) accounting for the frictional loss occurring during the transport of the drug through porous media. By means of fiber matrix theory, Curry (1984) initially derived an expression for the hindrance coefficient
where the reduction coefficient is computed by
With a mean fiber radius of \(\tilde{r}_\mathrm{f}=3.2\,\hbox {nm}\) (Formaggia et al. 2009), the values for the SES and media can be calculated.
Appendix 7: Diffusivity of sirolimus
No experimental data on the diffusivity of sirolimus in blood or blood plasma are available to the authors. Approximating sirolimus and paclitaxel as spheres, we estimate the average radius of sirolimus rescaling the radius of paclitaxel as a function of the molecular weight:
yielding
This result, combined with the Stokes–Einstein equation and the free plasma diffusivity of paclitaxel, then results in the adjusted diffusivity for sirolimus.
Rights and permissions
About this article
Cite this article
Bozsak, F., Chomaz, JM. & Barakat, A.I. Modeling the transport of drugs eluted from stents: physical phenomena driving drug distribution in the arterial wall . Biomech Model Mechanobiol 13, 327–347 (2014). https://doi.org/10.1007/s10237-013-0546-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10237-013-0546-4