
Abstract
Rare earth element (REE) patterns in natural water and geological samples provide information on previous changes in environmental conditions, such as redox changes and material cycles; however, quantitative analysis of REEs in these samples is complicated by the relatively low contents of REEs in such samples as well as mass interference from 135Ba16O and 137Ba16O in inductively coupled plasma mass spectrometry (ICP-MS) analyses. In this study, onsite solid-phase extraction and preconcentration methods for REEs using an iminobisacetic acid–ethylenediaminetriacetic acid chelate resin (Nobias Chelate PA1, Hitachi High-Tech Fielding) were adopted for the analyses. Standard reference materials (SPS-SW1 artificial surface water) and natural ground water and spring water samples were used to evaluate the methods. Using the chelate resin, background levels of REEs were found to be less than 0.3 ng L−1 and recovery rates (REEs, 1 ng L−1) were 97.9–106.7% for the artificial surface water. Ba contents were lower than the detection limit after extraction. Additionally, duplicate analyses were performed to check the reproducibility of the onsite extraction. The REE patterns in the natural water samples were in good agreement with those obtained using a previous method (the interference calibration method without solid-phase extraction). Therefore, onsite solid-phase extraction using the chelate resin was demonstrated to be a rapid and simple preparation technique for REE analyses.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The distribution and behavior of rare earth elements (REEs; i.e., lanthanoids and yttrium) and other chemical elements in natural environments are indicative of the current and previous chemical conditions and material sources in those environments (Wood et al. 1997; Och et al. 2014). Geochemical conditions, such as redox potential (oxic–anoxic conditions) and pH variations, play an important role in the material cycles that occur in limnological and geological environments. In addition, understanding geochemical conditions is crucial if we are to appropriately and safely use areas deep underground, e.g., to tap geothermal energy, to capture and store carbon dioxide (CCS), and to consider geological disposal (Dobashi and Shikazono 2008; Yamamoto et al. 2013).
Past geochemical conditions have been estimated by analyzing the iron (Fe), uranium (U), molybdenum (Mo), and REEs in natural environmental samples (e.g., ground water, sediments, rocks, and minerals; Iwatsuki et al. 2002; Munemoto et al. 2014; Siebert et al. 2015). Because carbonates (calcite, aragonite, and dolomite) are common minerals in the natural environment, geochemical proxies in carbonates have been used to estimate environmental changes and material cycles (Azmy et al. 2013). Relative amounts of Fe in carbonate samples have been used to infer past redox conditions, which can be calculated using a theoretical model based on the distribution coefficient between calcite deposits and dissolved ferrous ions (Arthur et al. 2006; Mizuno and Iwatsuki 2006). However, this theoretical model is limited to relatively low redox potentials and neutral pH conditions (JAEA 2015, 2016). REE contents show zigzag patterns (even-atomic-number elements are relatively dominant) based on the Oddo–Harkins rule. Cerium (Ce) and europium (Eu) anomalies are one of the most useful indicators of past redox conditions (Drake and Tullborg 2009; Feng et al. 2013). Additionally, chemical characteristics of trivalent REEs resemble those of artificial radioactive nuclides (trivalent actinides); therefore, the behavior of these nuclides in natural environments can be estimated using the REE distribution.
To understand past environmental changes and material cycles using REE patterns and other geochemical proxies in mineral samples, it is necessary to determine the distribution of RREs and their behavior in the original solution (ground water in this case). REE preconcentration and separation processes are required to analyze ground water samples because of the extremely low concentrations (less than ca. 0.1 ng L−1, Munemoto et al. 2015) of REEs in such samples. Usefully, the recent development of inductively coupled plasma mass spectrometry (ICP-MS) techniques has enabled parts per trillion (ppt, 1 ng L−1 or 1 ng kg−1) or sub-ppt-level REE measurements to be performed for natural and artificial samples.
However, when performing high-sensitivity REE measurements using ICP-MS, mass interference from barium oxide (135Ba16O and 137Ba16O) needs to be cleared and calibrated, particularly when carrying out small-amount europium (151Eu and 153Eu) quantification. To remove these oxides during ICP-MS measurements, helium collision, a dynamic reaction cell with ammonia or methane gas, or a cool plasma technique can be adopted for large matrix samples (Ogawa et al. 2010; Fialho et al. 2011); however, the application of these techniques results in relatively low sensitivity for each target ion in the ICP-MS measurements (~μg L−1 or ~mg L−1). High-sensitivity analyses of ng L−1 levels are necessary for liquid-phase samples in limnological and geological research. Thus, REE extraction has been performed using organic solvents, the iron co-precipitation method, an ion exchange resin, and an iminobisacetic acid chelating resin (Kikawada et al. 2013; Bayon et al. 2015; Munemoto et al. 2015). However, these techniques are unable to remove alkaline earth metals, which cause mass interference in ICP-MS measurements.
A wide variety of preparation methods for REEs in natural water have been reported (Fisher and Kara 2016). Commercial iminobisacetic acid–ethylenediaminetriacetic acid chelate resin has no affinity for alkali metals and alkaline earth metals (Sakamoto et al. 2006; Yamamoto et al. 2007), so this chelate resin was examined as a possible means for REE preconcentration and the elimination of alkali metals and alkaline earth metals from groundwater samples. This chelate resin has previously been used to preconcentrate micronutrients (Fe, Cu, Zn, Cd) in seawater and salt water samples (Furusho et al. 2008; Tsuneto et al. 2009; Yamazaki et al. 2009; Zhu et al. 2010; Aosai et al. 2014); however, the application of this resin to REE analyses of ground water samples has rarely been reported. Natural ground water has various chemical components and physical parameters, and REE contents in ground water are extremely low (less than ca. 0.1 ng L−1).
Additionally, onsite solid-phase extraction techniques using a chelate resin could be rapid and simple because water properties, such as redox potential, may be quickly changed after sampling by oxidation (this may depend on the sample; Iida et al. 2009; Sanada et al. 2013; Saitoh et al. 2013). Such extraction techniques could also be used to preconcentrate a large amount of ground water (more than 1 L) at the sampling site. In particular, extraction in this manner could be useful for ground water sampling in deep and narrow caves. In the study reported in the present paper, background, recovery, and reproducibility tests of standard reference materials (artificial surface water) were performed using a chelate resin in order to establish onsite solid-phase extraction techniques. This study is the first to evaluate onsite solid-phase extraction using an iminobisacetic acid–ethylenediaminetriacetic acid chelate resin for ground water and spring water.
Materials and analytical methods
Chelate resin
An iminobisacetic acid–ethylenediaminetriacetic acid chelate resin (Nobias Chelate PA1 syringe type, 45–90 μm resin diameter, 250 mg, Hitachi High-Tech Fielding) was used for solid-phase extraction in this study. Solid-phase extraction by the chelate resin was performed according to the steps shown the analytical flowchart in Fig. 1. First, the chelate resin was cleaned and swelled with 10 mL ethanol (soaked for 5 min) before being rinsed with 3 M HNO3 (10 mL, for analysis of poisonous metals, Wako Pure Chemical Industries) and with ultrapure water (20 mL, 18.2 MΩ cm, Milli-Q Integral 5 water purification system, Merck Millipore). Then, 0.1 M ammonium acetate (10 mL, JIS Special Grades, Wako Pure Chemical Industries) was added to adjust the pH of the chelate resin for conditioning before sample loading. Finally, samples (20 or 100 mL, with pH adjusted to 5–6 using an ammonium acetate solution) were loaded on the chelate resin.

Flowchart of the quantitative analysis of REEs and other elements in artificial surface water (SW-1, Spectrapure Standards AS, 100 mL) and natural water samples (20 or 100 mL) obtained by solid-phase extraction using the chelate resin
Alkali metals and alkaline earth metals are not trapped on the chelate resin, so they could be flushed from the chelate resin using ultrapure water (10 mL) following sample loading. REEs and other elements were recovered from the chelate using 3 M HNO3 (3 mL). The REE contents in residues obtained using an extra elution were also checked (3 M HNO3, 3 mL, Fig. 1). The flow rate of the eluent was ca. 3 mL/min (natural flow). The volume was measured using a manual pipetting system (Finnpipette, Thermo Fisher Scientific) calibrated to a 10 µg scale electric balance. Each eluate (one for REE quantification and one for residues; Fig. 1) was diluted to 10 mL using ultrapure water and acid was added to a concentration of 5 v/v% HNO3 before instrumental analyses.
Instruments
Concentrations of REEs and other elements were measured three times for each sample using an ICP-MS system (7700x, Agilent Technologies) in the Toki Research Institute of Isotope Geology and Geochronology, JAEA. Flow rates of nebulizer gas, plasma gas, and auxiliary gas (pure argon, 99.999%) were 1.02, 16, and 1.2 mL/min, respectively, and the RF power for the ICP-MS was 1500 W (hot plasma conditions). Nickel skimmer and sampling corns were used for the ICP-MS analyses. The oxide interference (140Ce16O+/140Ce+) and doubly charged atomic ion (Ce2+/Ce+) ratios were approximately 1% under these conditions. REEs, alkali metals, alkaline earth metals, heavy metals (Co, Cu, Mo, Cd, Th, U) and other elements (V, Mn, As) were measured in the study (Table 1). Scandium (Sc) could not be measured under these conditions because of mass interference from molecules such as 13C16O16O, 12C16O17O, and 44Ca1H. Concentrations of the elements were calculated using calibration standards (0.1, 0.2, 0.5, 1, 2, 5, 10, 50, 100 ng L−1 for mixed standard solutions: XSTC-1 and XSTC-331, SPEX). The correlation coefficient values of the calibration curves of the REEs were more than 0.99. An internal standard of 115In (SPEX) was used for quantification by ICP-MS. The standard deviation of triplicate ICP-MS measurements of the REEs in the mixed standard solution was within ca. 5% and 10% (±1σ) at 10 and 1 ng L−1, respectively.
Blank, recovery, and reproducibility tests
Blank tests of ultrapure water (n = 5) before and after extraction by the chelate resin were performed. For the REE recovery tests, standard reference materials with 1–100 and 0.1–10 ng L−1 (Table 2) were measured using ICP-MS (n = 4, 100 mL, SPS-SW1 artificial surface water, Spectrapure Standards AS). To evaluate the onsite solid-phase extraction by the chelate, ground water samples from a cave in Gifu (GJ01a, GJ01b and GJ02; 35.7°N, 137.0°E, October 20, 2014) and spring water samples from Yamanashi (MS05 and MS13; 35.9°N, 138.5°E, October 27, 2014) in the middle of Japan were analyzed by ICP-MS before and after extraction. The GJ01a and GJ01b ground water samples were obtained from the same points for reproducibility. The GJ02 samples were taken from another point close to the GJ01 sampling point.
The chelate resin was cleaned and conditioned in the laboratory before sampling. Natural water samples were filtered (pore size 0.45 μm, membrane filter, DISMIC 25CS045AN, ADVANTEC). For the solid-phase extraction procedure, the sample volume was 20 mL (GJ01 and GJ02) or 100 mL (MS05 and MS13). The REEs and other elements were trapped by the chelate just after filtration at the sampling sites, and the elution processes and measurements were performed in the Toki Research Institute of Isotope Geology and Geochronology. The pH values of the original solution for GJ01, GJ02, MS05, and MS13 were 7.60, 7.63, 6.26, and 6.73, respectively (measured using a portable pH meter, D-54SE, Horiba). The samples were acidified using concentrated HNO3 (for analysis of poisonous metals, Wako Pure Chemical Industries) after filtration at the sampling sites by the previously reported method for comparison. The acidified samples were diluted and the internal standard was added for quantification by ICP-MS. Mass interference in the REEs analyses was calibrated using a barium sole standard solution (SPEX).
Results and discussion
Blank test
Average background counts of REEs in ultrapure water before the extraction ranged from 5 to 59 counts per second (cps), and the background equivalent concentrations (BECs) ranged from <0.1 to 0.3 ng L−1 in the study (Table 1). The BEC values for Mo, Th, and U were less than 0.1 ng L−1. The BEC values were sufficiently low to allow the quantification of natural land water samples using ICP-MS when compared with previously obtained values (Sohrin et al. 2008; Ogawa et al. 2010; Watanabe et al. 2014). Relatively high background counts were observed for elements with low atomic numbers (55Mn, 59Co, 63Cu, and 65Cu); this was caused by mass interference from argon molecules in the carrier gas and other sources in ICP-MS (e.g., 40Ar14N1H, 40Ar18O1H, 39Ar12C12C, 40Ar12C13C, 39Ar12C14N).
Results of the blank test of REEs and other elements in ultrapure water (100 mL) after solid-phase extraction using the chelate resin are listed in Table 1. Counts for most of the REEs after extraction were lower than the detection limit. On the other hand, relatively high background values of lanthanum (La) and cerium (Ce) of up to 0.3 ng L−1 were observed, and those of yttrium (Y) were 0.3–0.5 ng L−1; however, these background values had no effect on the quantification results obtained by ICP-MS because the natural abundances of Y, La, and Ce are higher than those of other REEs in many cases. The background counts were stable, and the detection limits and BEC values were sufficiently low for REEs and other elements to permit quantification by ICP-MS analysis in the study (Table 1).
Recovery test
Recovery rates of REEs in the artificial surface water (SPS-SW1) by solid-phase extraction using the chelate resin ranged from 97.9 to 106.7% and from 91.6 to 127.4% for 1 and 0.1 ng L−1 REEs, respectively (Table 2). The REE levels in the residues (3–6 mL HNO3, Fig. 1) were lower than the corresponding detection limits. These results indicate that 3 mL of 3 M HNO3 was sufficient to achieve complete REE recovery from the chelate resin. Coefficient variations across the four analyses conducted for the elements in the 1 ng L−1 REEs solution were up to 4.7%; this value is within the analytical uncertainty for ICP-MS (see “Materials and analytical methods”). The coefficient variations of the elements in the 0.1 ng L−1 REEs solution were relatively large (from 1.9 to 14.9%) compared with those for the 1 ng L−1 solution; the uncertainties were all within 10% except that for neodymium (Nd). These values could be sufficient to consider geochemical studies using REE patterns on a logarithmic scale (Wyndham et al. 2004; Sandström et al. 2009; Bourdin et al. 2011; Maskenskaya et al. 2015).
The recovery rates of Mo and U were also close to 100% in the study (initial contents: 20 and 1 ng L−1, respectively; Table 2); however, the Mo recovery rate in the solution with 2 ng L−1 could not be observed because of relatively high background values in the ICP-MS (Tables 1, 2). Regarding the other elements (Mn, Co, Cu, and Cd), high recovery rates of 99.8–109.2% on average were obtained in this study. The recovery rate of As was relatively low and unstable with uncertainties of up to ca. 20%; this could be caused by low affinity for the chelate resin and high pH sensitivity of the extraction by the chelate. Therefore, we should exclude As as a target in this method because of its low recovery rate. For the REEs, pH values in the range of 5–6 were reasonable, as demonstrated by the high recovery rates seen in this study. High recovery rates with pH values in the range 5–6 have also been observed by Sohrin et al. (2008). Therefore, appropriate pH adjustment could be necessary to extract target elements.
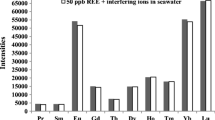
Contents of alkali and alkaline earth metals (Rb, Sr, Cs, and Ba) were lower than the detection limits and were removed adequately by extraction using the chelate resin. Therefore, Ba oxide interference could not occur under these conditions for the REE analyses; however, Ba contents were 100 times higher than those of the REEs in the original standard solution. These results indicate that the chelate resin could be useful, particularly for preparation in the analysis of REEs in large-matrix samples.
Onsite solid-phase extraction
The REEs in two ground water samples taken from same point (GJ01a and GJ01b) were extracted by the chelate from the sampling site for reproducibility. The differences in REE concentrations between the two ground water samples were within the analytical uncertainty of the ICP-MS measurement (Fig. 2). The reproducibility of the analysis of REEs at ng L−1 and sub ng L−1 levels was confirmed for the natural ground water samples in the study by onsite solid-phase extraction using the chelate resin.

Reproducibility of the REE levels in ground water (GJ01) from Gifu, middle Japan, after onsite solid-phase extraction using chelate resin
The REE patterns in GJ01 (average values of GJ01a and GJ01b, same sampling point), GJ02, MS05, and MS13 after onsite solid-phase extraction using the chelate resin were obtained, and those obtained by previous methods are also shown for comparison in Figs. 3 and 4. For ground water samples from the cave (GJ01 and GJ02), the REE contents were approximately 0.1–5 ng L−1, and typical REE patterns were observed, with enrichment in lighter REEs. Although some REE contents were lower than the detection limits for the previous method (Figs. 3, 4), most of the REE contents could be obtained by onsite solid-phase extraction using the chelate resin; an exception was samarium (Sm) in GJ02 (less than the detection limit). Additionally, the order of the REE contents and the patterns obtained by the chelate were similar to the data obtained from the previous method (Figs. 3, 4). The use of the chelate resin for REE preconcentration in the natural samples facilitated the quantification of extremely low amounts of REEs (less than 0.1 ng L−1), particularly for heavy REEs such as Tm, Yb, and Lu.

REE concentrations in a, b ground water from Gifu and c, d spring water samples from Yamanashi, middle Japan, as obtained by onsite solid-phase extraction using chelate resin (filled circles) and by the previous method (open diamonds; before chelate resin extraction, see the “Materials and analytical methods” section)

REE patterns in a, b ground water from Gifu and c, d spring water samples from Yamanashi, middle Japan, as obtained by onsite solid-phase extraction using chelate resin (filled circles) and by the previous method (open diamonds; before chelate resin extraction, see the “Materials and analytical methods” section)
The REE contents in the spring water samples (MS05 and MS13, Fig. 3c, d) varied widely, from 0.1 to 100 ng L−1. Relative enrichment of heavy REEs were found in MS05, and high REE contents of up to 106 ng L−1 were observed in MS13. The REE pattern in MS13 as determined by the extraction was also in good agreement with that obtained using the previous method (Fig. 4d). The chelate resin was available for samples with a wide range of REE contents. Additionally, the negative anomaly trend for Eu was also observed in the MS13 spring water samples before and after extraction by the chelate; this possibly indicates redox conditions and/or material sources in the environment. Therefore, onsite solid-phase extraction using the chelate resin is a useful preparation method for quantification by ICP-MS.
Concentrations of Ba ranged from ca. 6,800 to 31,900 ng L−1 in the original water samples before the extraction in the study. After the extraction, the levels of alkali and alkaline earth metals were decreased by the chelate resin to ca. 20 ng L−1 in the ground water samples. Thus, Ba interference in the REE analyses was sufficiently reduced by the chelate resin extraction from the natural samples. Although Ba mass interference calibration was employed for quantification in previous methods, the uncertainty in the Ba count (μg–mg L−1 level) obtained by ICP-MS could affect the calibrated values of small amounts of Eu (sub ng L−1 level). Therefore, compared with the previous method, quantification without mass-interference calibration by the Ba elimination technique using the chelate resin could be a better and easier method of obtaining a high-precision data set. Because the sample volume of ground water is usually limited, the elimination of alkali and alkaline earth metals is a more valuable technique than preconcentration in this case. These results indicate that onsite solid-phase extraction using a chelate could be a stable, unique, and useful technique for preparation REE for ICP-MS analysis.
Conclusions
The onsite solid-phase extraction of REEs using an iminobisacetic acid–ethylenediaminetriacetic acid chelate resin for natural land water samples was established as follows:
-
1.
A blank test of REEs after extraction by the chelate indicated that background REE levels were sufficiently low (<0.1–0.3 ng L−1) to permit ICP-MS analyses
-
2.
Recovery rates of REEs by the chelate were more than 97.9 ± 2.1 and 86.4 ± 6.7% in the artificial surface water samples (SW-1) with initial REE contents of 1 and 0.1 ng L−1, respectively
-
3.
The interfering element (Ba) was completely removed by the chelate (Ba contents were less than the detection limit in SW-1) for the REE analyses by ICP-MS
-
4.
The reproducibility of the REE measurements was demonstrated by performing duplicate analyses of ground water samples
-
5.
Typical REE patterns were observed for ground and spring water samples upon performing onsite solid-phase extraction using the chelate resin.
Therefore, this could potentially be a standard method for REE analyses in natural land water samples.
References
Aosai D, Yamamoto Y, Mizuno T, Ishigami T, Matsuyama H (2014) Size and composition analyses of colloids in deep granitic groundwater using microfiltration/ultrafiltration while maintaining in situ hydrochemical conditions. Colloid Surf A 461:279–286
Arthur RC, Iwatsuki T, Sasao E, Metcalfe R, Amano K, Ota K (2006) Geochemical constraints on the origin and stability of the Tono uranium deposit, Japan. Geochem Explor Environ Anal 6:1–16
Azmy K, Lavoie D, Wang Z, Brand U, Al-Aasm I, Jackson S, Girard I (2013) Magnesium-isotope and REE compositions of Lower Ordovician carbonates from eastern Laurentia: implications for the origin of dolomites and limestones. Chem Geol 356:64–75
Bayon G, Toucanne S, Skonieczny C, Andre L, Bermell S, Cheron S, Dennielou B, Etoubleau J, Freslon N, Gauchery T, Germain Y, Jorry SJ, Meenot G, Monin L, Ponzevera E, Rouget M-L, Tachikawa K, Barrat JA (2015) Rare earth elements and neodymium isotopes in world river sediments revisited. Geochim Cosmochim Acta 170:17–38
Bourdin C, Douville E, Genty D (2011) Alkaline-earth metal and rare-earth element incorporation control by ionic radius and growth rate on a stalagmite from the Chauvet Cave, Southeastern France. Chem Geol 290:1–11
Dobashi R, Shikazono N (2008) Geochemical study of rare earth elements in carbonate minerals in sedimentary rocks around Tono uranium deposit, central Japan—an example of natural analogue study of geological disposal of high-level nuclear waste. Chikyukagaku (Geochemistry) 42:79–98 (in Japanese with English abstract)
Drake H, Tullborg E-L (2009) Paleohydrogeological events recorded by stable isotopes, fluid inclusions and trace elements in fracture minerals in crystalline rock, Simpevarp area, SE Sweden. Appl Geochem 24:715–732
Feng D, Lin Z, Bian Y, Chen D, Peckmann J, Bohrmann G, Roberts HH (2013) Rare earth elements of seep carbonates: indication for redox variations and microbiological processes at modern seep sites. J Asian Earth Sci 65:27–33
Fialho LL, Pereira CD, Nobrega JA (2011) Combination of cool plasma and collision-reaction interface for correction of polyatomic interferences on copper signals in inductively coupled plasma quadrupole mass spectrometry. Spectrochim Acta B 66:389–393
Fisher A, Kara D (2016) Determination of rare earth elements in natural water samples—a review of sample separation, preconcentration and direct methodologies. Anal Chim Acta 935:1–29
Furusho Y, Ono M, Yamada M, Ohashi K, Kitade T, Kiriyama K, Ohta S, Inoue Y, Motomizu S (2008) Advanced solid phase extraction for inorganic analysis and its applications. Bunseki Kagaku 57:969–989 (in Japanese with English abstract)
Iida Y, Kimura Y, Yamaguchi T, Ueda M, Tanaka T, Nakayama S (2009) Sorption distribution coefficients of selenium on a sandy mudstone under reducing conditions of underground. J Nucl Fuel Cycle Environ 15(2):57–67 (in Japanese with English abstract)
Iwatsuki T, Satake H, Metcalfe R, Yoshida H, Hama K (2002) Isotopic and morphological features of fracture calcite from granitic rocks of the Tono area, Japan: a promising palaeohydrogeological tool. Appl Geochem 17:1241–1257
JAEA (2015) 2014 Annual report on METI (Ministry of Economy, Trade and Industry) R&D supporting program for developing geological disposal technology. JAEA, Tokyo (in Japanese)
JAEA (2016) 2015 Annual report on METI (Ministry of Economy, Trade and Industry) R&D supporting program for developing geological disposal technology. JAEA, Tokyo (in Japanese)
Kikawada Y, Fukai M, Oi T (2013) Specific REE patterns observed in sulfurous hot springs from a hydrothermal alteration area in Manza, Japan. Proce Earth Planet Sci 7:428–431
Maskenskaya OM, Drake H, Mathurin FA, Åström ME (2015) The role of carbonate complexes and crystal habit on rare earth element uptake in low-temperature calcite in fractured crystalline rock. Chem Geol 391:100–110
Mizuno T, Iwatsuki T (2006) Long-term stability of geochemical environment at deep underground. Case study of minor elements in carbonate minerals. Chikyukagaku (Geochemistry) 40:33–45 (in Japanese with English abstract)
Munemoto T, Ohmori K, Iwatsuki T (2014) Distribution of U and REE on colloids in granitic groundwater and quality-controlled sampling at the Mizunami Underground Research Laboratory. Prog Earth Planet Sci 1:28
Munemoto T, Ohmori K, Iwatsuki T (2015) Rare earth elements (REE) in deep groundwater from granite and fracture-filling calcite in the Tono area, central Japan: prediction of REE fractionation in paleo- to present-day groundwater. Chem Geol 417:58–67
Och LM, Müller B, Wichser A, Ulrich A, Vologina EG, Sturm M (2014) Rare earth elements in the sediments of Lake Baikal. Chem Geol 376:61–75
Ogawa Y, Yamasaki S, Tsuchiya N (2010) Application of a dynamic reaction cell (DRC) ICP-MS in chromium and iron determination in rock, soil and terrestrial water samples. Anal Sci 26:867–872
Saitoh Y, Iijima A, Kimura S, Kozawa K (2013) Preliminary study on evaluation of rare earth elements in hot spring water as a geochemical indicator intended to understand hot spring water flow. J Hot Spring Sci 63:141–157 (in Japanese with English abstract)
Sakamoto H, Yamamoto K, Shirasaki T, Inoue Y (2006) Pretreatment method for determination of trace elements in seawater using solid phase extraction column packed with polyamino-polycarboxylic acid type chelating resin. Bunseki Kagaku 55:133–139 (in Japanese with English abstract)
Sanada T, Nagashima H, Matsumoto G-I (2013) Geochemical features and formation mechanism of chemical components in the Goshikinuma water system of Urabandai, Fukushima prefecture in Japan. J Hot Spring Sci 63:13–27 (in Japanese with English abstract)
Sandström B, Tullborg E-L, Larson SA, Page L (2009) Brittle tectonothermal evolution in the Forsmark area, central Fennoscandian Shield, recorded by paragenesis, orientation and 40Ar/39Ar geochronology of fracture minerals. Tectonophysics 478:158–174
Siebert C, Pett-Ridge JC, Opfergelt S, Guicharnaud RA, Halliday AN, Burton KW (2015) Molybdenum isotope fractionation in soils: influence of redox conditions, organic matter, and atmospheric inputs. Geochim Cosmochim Acta 162:1–24
Sohrin Y, Urushihara S, Nakatsuka S, Kono T, Higo E, Minami T, Norisuye K, Umetani S (2008) Multielemental determination of GEOTRACES key trace metals in seawater by ICPMS after preconcentration using an ethylenediaminetriacetic acid chelating resin. Anal Chem 80:6267–6273
Tsuneto A, Suzuki Y, Furusho Y, Furuta N (2009) Development of the determination method of rare earth elements in seawater by ICP-MS with an on-line preconcentration column of improved iminodiacetate resin and its application to Tokyo Bay seawater. Bunseki Kagaku 58:623–631 (in Japanese with English abstract)
Watanabe T, Tsuchiya N, Yamasaki S, Yamada R, Hirano N, Okamoto A, Nara FW, Tohoku Tsunami Sediment Research Group (2014) Seawater-leaching testing for arsenic and heavy metals in tsunami deposits produced by the 2011 off the Pacific coast of Tohoku earthquake, northeastern Japan. Chigaku Zasshi 123:835–853 (in Japanese with English abstract)
Wood SA, van Middlesworth P, Gibson P, Ricketts A (1997) The mobility of the REE, U and Th in geological environments in Idaho and their relevance to radioactive waste disposal. J Alloys Compounds 249:136–141
Wyndham T, Mcculloch M, Fallon S, Alibert C (2004) High-resolution coral records of rare earth elements in coastal seawater: biogeochemical cycling and a new environmental proxy. Geochim Cosmochim Acta 68:2067–2080
Yamamoto K, Sakamoto H, Yonetani A, Shirasaki T (2007) Determination of trace metals in salt and sea water by means of ICP-AES and MIP-MS with preconcentration using the polyamino-polycarboxylic acid type chelating resin solid phase extraction. Bull Soc Sea Water Sci 61:260–267 (in Japanese)
Yamazaki, M, Kato, S, Tsukada, S, Yoshioka, O (2009) Analysis of trace metal elements in environmental waters by solid-phase extraction using iminobisacetic acid–ethylenediaminetriacetic acid chelating resin. Annu Rep Mie Prefect Health Environ Res Inst 11:108–116 (in Japanese)
Zhu Y, Itoh A, Umemura T, Haraguchi H, Inagaki K, Chiba K (2010) Determination of REEs in natural water by ICP-MS with the aid of an automatic column changing system. J Anal At Spectrom 25:1253–1258
Acknowledgements
This work was partly supported by a METI (Ministry of Economy, Trade and Industry of Japan) program for the research and development of technology relating to geological disposal. The authors express their gratitude to the Chubu branch office of the Hitachi High-Tech Fielding Corporation. N. Takamura, Y. Shorin, and an anonymous reviewer are acknowledged for their helpful comments and suggestions for improving the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling Editor: Yoshiki Sohrin.
Rights and permissions
About this article

Cite this article
Watanabe, T., Saito-Kokubu, Y., Murakami, H. et al. Onsite chelate resin solid-phase extraction of rare earth elements in natural water samples: its implication for studying past redox changes by inorganic geochemistry. Limnology 19, 21–30 (2018). https://doi.org/10.1007/s10201-017-0513-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10201-017-0513-3