
Abstract
Background
Hereditary hypomagnesemia is difficult to diagnose accurately because of its rarity and the variety of causative genes. We established a flowchart for identifying responsible genes for hypomagnesemia, and we confirmed its diagnostic efficacy in patients with suspected inherited hypomagnesemia.
Methods
We established a flowchart and applied it to five index cases with suspected inherited hypomagnesemia. Direct sequence analysis was used to detect the causative gene variants in four cases, and targeted sequencing analysis using next-generation sequencing (NGS) of all causative genes for hypomagnesemia was used in one.
Results
Expected pathogenic variants were detected in the HNF1B, TRPM6, CLDN16, CASR, or SLC12A3 gene in all five cases. The results of all genetic analyses were consistent with the clinical diagnostic results using the flowchart.
Conclusions
Accurate genetic diagnosis is crucial for estimating the prognosis, detecting complications in organs other than the kidneys, and for directing genetic counseling. The developed flowchart for identifying responsible genes for hypomagnesemia was useful for diagnosing inherited hypomagnesemia. In addition, NGS analysis will help to resolve clinical difficulties in making an accurate diagnosis and thus improve the diagnostic strategy for inherited hypomagnesemia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Magnesium (Mg2+) is a cofactor for a group of enzymes and transporters, and it also plays an essential role in the synthesis of nucleic acids and proteins [1]. Hypomagnesemia is defined as a serum Mg2+ level < 1.7 mg/dL (<0.7 mmol/L) [2]. Patients with hypomagnesemia suffer from nonspecific symptoms such as depression, tiredness, muscle spasms, or muscle weakness [1], while severe Mg2+ depletion (<0.4 mmol/L) may lead to cardiac arrhythmias, tetany, or seizures [1]. Hypomagnesemia can be caused by many triggers, including alcohol abuse, chronic diabetes, drugs, or eclamptic seizures [3], while genetic hypomagnesemia can be distinguished from Mg2+ deficiency arising from other causes [1]. However, the genetic causes of hypomagnesemia are also heterogeneous and comprise both recessive and dominant disorders (Table 1) [2].
Hereditary hypomagnesemia may be difficult to diagnose, because it is a relatively rare disorder and exhibits a variety of clinical presentations. We established a flowchart for identifying responsible genes for hypomagnesemia (Fig. 1) and demonstrated its efficacy in five index cases.

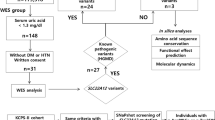
Flowchart for identifying responsible genes for hypomagnesemia. The flowchart required the following clinical data for identifying responsible genes for hypomagnesemia: (1) congenital anomalies of the kidney and urinary tract (CAKUT) or family history of maturity-onset diabetes of the young (MODY); (2) hypokalemia and metabolic alkalosis (Gitelman-like); (3) hearing loss; (4) hypo or hypercalciuria; (5) hypoparathyroidism and hypocalcemia; and (6) ocular anomalies
Materials and methods
Patients
We investigated five index cases with hypomagnesemia (Table 2). Clinical and laboratory data and pathological findings were obtained from medical records.
Genetic analysis
Genomic DNA was isolated from peripheral blood leukocytes from the patients and their family members using the Quick Gene Mini 80 system (Wako Pure Chemical Industries, Ltd., Tokyo, Japan) according to the manufacturer’s instructions. Direct sequencing or next-generation sequencing (NGS) was conducted for genes responsible for inherited hypomagnesemia. NGS samples were prepared using a HaloPlex target enrichment system kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer’s instructions. The inherited hypomagnesemia-responsible genes CLCNKB, BSND, SLC12A3, CASR, KCNJ10, CLDN16, CLDN19, FXYD2, EGF, TRPM6, KCNA1, CNNM2, and HNF1B were screened by targeted sequencing.
Ethical considerations
All procedures were approved by the Institutional Review Board (IRB) of Kobe University Graduate School of Medicine and in accordance with the Helsinki Declaration of 1975, as revised in 2000 (IRB number: 301). Informed consent was obtained from all index patients or their parents.
Results
Patients
Patient 1 was a 2-year-old boy who was found to have low serum Mg2+ by chance at a regular visit for observation of hypoplastic kidney. He had been born at 36 weeks to unrelated parents after a hydramniotic pregnancy, with a birth weight of 2250 g. His father has an isolated renal cyst and diabetes mellitus, his older sister had died immediately after birth because of Potter’s sequence, and his younger brother also had renal hypoplasia. Laboratory test results are shown in Table 2. According to the flowchart, HNF1B gene mutation was suspected based on the clinical presentation in this family with CAKUT and diabetes, and direct sequencing of this gene was conducted.
Patient 2 was a 26-year-old man. He had been born at 39 weeks to unrelated parents after an uncomplicated pregnancy, with a birth weight of 2700 g. He presented with recurrent seizures at 1 month old, and although laboratory tests revealed hypomagnesemia and hypocalcemia, no further examination was conducted at that time. His younger brother underwent blood tests at 4 days old because of his family history, which also revealed hypomagnesemia. The results of laboratory tests for the index patient performed at 1 month old are shown in Table 2. He required intravenous Mg2+ two or three times a week since the initial diagnosis. This patient had no ocular involvement. His and his brother’s current laboratory data are shown in Table 2. We conducted targeted sequencing analysis using NGS, including hereditary hypomagnesemia-causative genes. Based on the clinical presentation of hypomagnesemia and hypocalcemia and intact parathyroid hormone (iPTH) suppression without CAKUT, hypokalemia, or hypercalciuria, this family was suspected to have TRPM6, EGF/EGFR, or FAM111A variants.
Patient 3 was a 6-month-old girl who was born to unrelated parent at 38 weeks after an uncomplicated pregnancy, with a birth weight of 2680 g. Low serum Ca2+ and Mg2+ were detected by chance during a bout of viral gastroenteritis. Her asymptomatic elder sister was tested after the diagnosis of Patient 3 and was also found to have hypomagnesemia. Laboratory test results for both sisters are shown in Table 2. She had no ocular involvement. The clinical presentation of hypercalciuria without CAKUT, hypokalemia, iPTH suppression, or ocular abnormality suggested CLDN16 mutation, and direct sequencing of this gene was conducted.
Patient 4 was a 33-year-old woman, whose case has been reported previously [4]. Low serum Ca2+ was detected by chance in the absence of any clinical signs, and she was diagnosed with hypocalcemia and hypoparathyroidism at the age of 6 years. She showed hypokalemia, metabolic alkalosis, and short stature at the age of 12, and was administered growth hormone therapy for 3 years. Hypomagnesemia was detected when she was 25 years old. She developed bilateral renal calcification and mild impairment of kidney function at the age of 33 years. She had been prescribed active vitamin D3 since the age of 6 years, but this was tapered off after a diagnosis of ADH1. The results of laboratory tests performed at her initial visit to our hospital are shown in Table 2. From the clinical presentation of hypokalemia, hypercalciuria, iPTH suppression, and hypocalcemia without CAKUT or hearing loss, she was suspected to be have a CASR mutation, and direct sequencing of this gene was conducted.
Patient 5 was an 8-year-old girl who had been born at 40 weeks to unrelated parents after an uncomplicated pregnancy, with a birth weight of 3246 g. She had no remarkable family or past history. However, low serum Mg2+ was detected by chance when she was affected by viral gastroenteritis. Her laboratory test results are shown in Table 2. She had no significant inner ear involvement. Based on her clinical presentation of hypokalemia and hypocalciuria, without hearing loss, she was suspected to have an SLC12A3 mutation.
Flowchart
The flowchart was established according to the following reports of inherited hypomagnesemia. HNF1B is a causative gene of CAKUT and early onset diabetes, and MODY5 that is also caused by HNF1B variants is frequently accompanied by hypomagnesemia [5–7]. Patients with FXYD2, KCNA1, CNNM2, or PCBD1 variants show hereditary hypomagnesemia with hypo- to normocalciuria [8–12]. Hypomagnesemia with secondary hypocalcemia (HSH) caused by TRPM6 variants is associated with hypo- to normocalciuria with serum iPTH suppression [13–15]. Kenny-Caffey syndrome type 2 is a rare condition caused by FAM111A variants. It is characterized by cortical thickening and medullary stenosis of tubular bones, delayed closure of the anterior fontanelle, eye abnormalities, hypoparathyroidism and hypocalcemia accompanied by hypomagnesemia, and hypo- to normocalciuria [16]. ADH1/type V Bartter syndrome caused by CASR variants also shows iPTH supression and hypocalcemia [17–19]. Patients with EGF or EGFR variants may share similar pathophysiology with HSH, because EGF increases TRPM6 activity and surface expression [20, 21]. Familial hypomagnesemia with hypocalcemia and nephrocalcinosis (FHHNC) caused by CLDN16 or CLDN19 variants is associated with hypercalciuria, while CLDN19 variants are associated with ocular impairment [22–24]. Hypomagnesemia accompanied by hypokalemia and metabolic alkalosis (Gitelman-like) indicates the possibility of Bartter/Gitelman syndrome-associated disorders. Both type III Bartter syndrome caused by variants in CLCNKB and Gitelman syndrome caused by variants in SLC12A3 are usually accompanied by hypomagnesemia [25–28]. Pathogenic variants in the BSND gene cause type IV Bartter syndrome with sensorineural deafness and occasionally hypomagnesemia [2, 29]. Pathogenic variants in KCNJ10 also cause hypokalemic and hypomagnesemic tubulopathy with hearing loss (EAST syndrome), and this tubulopathy is identical to that seen in Gitelman syndrome [30].
The flowchart required the following clinical data for identifying responsible genes for hypomagnesemia: (1) congenital anomalies of the kidney and urinary tract (CAKUT) or family history of maturity-onset diabetes of the young (MODY); (2) hypokalemia and metabolic alkalosis (Gitelman-like); (3) hearing loss; (4) hypo- or hypercalciuria; (5) hypoparathyroidism and hypocalcemia; and (6) ocular anomalies (Fig. 1).
Genetic analysis
All genetic test results are shown in Tables 2 and 3. The results of all genetic analyses were consistent with the clinical diagnostic results obtained using the flowchart (Fig. 1). Truncating variants were detected in Patients 1 and 3. A splicing variant was detected in Patient 2. Missense variants were detected in Patients 4 and 5, both of which were predicted as pathogenic by in silico analysis. In addition, variants in Patients 1, 2, 4 and 5 have been reported as pathogenic (Table 3) [31–34].
Discussion
This study demonstrated the validity of our flowchart in five index cases of inherited hypomagnesemia. The flowchart is necessarily complicated because of the genetic heterogeneity of the condition. However, an accurate clinical diagnosis is important in terms of the kidney prognosis, the presence of complications other than hypomagnesemia, and for conducting genetic counseling. Recent developments in genetic techniques mean that it is possible to make a genetic diagnosis using NGS, even in the absence of an accurate clinical diagnosis, as in Patient 2. However, a clinical diagnosis remains important, because NGS is not always available. A diagnostic flowchart thus provides a highly useful tool for clinicians.
Heterozygous variants in HNF1B result in multi-system disorders and are the most common monogenic cause of CAKUT, occurring in 10–30% of CAKUT patients in the prenatal period [7]. HNF1B is also a causative gene of early onset diabetes, MODY5 [6]. In addition, an initial report showed that hypomagnesemia occurred in up to 50% of affected patients [5]. The presence of CAKUT with hypomagnesemia and a family history of diabetes provided a useful clue to a potential HNF1B mutation in Patient 1. Serum Mg2+ levels should be monitored in patients with CAKUT as an indicator of potential HNF1B mutations, while a family history of early onset diabetes is also a clue for the detection of HNF1B variants, as in Patient 1.
Homozygous or compound heterozygous variants in TRPM6 cause HSH, which is a relatively rare autosomal recessive disease [15]. Affected individuals present in early infancy with seizures caused by the severe hypocalcemia and hypomagnesemia [15]. HSH is sometimes misdiagnosed as primary hypoparathyroidism because of its initial presenting symptoms of hypocalcemia and concomitant low or inappropriate normal parathyroid hormone (PTH) caused by hypomagnesemia, which blocks the release of PTH and decreases sensitivity to circulating PTH in target organs [13]. Some HSH patients can be managed by Mg2+ supplementation without a genetic diagnosis, as in Patient 2. HSH is a relatively rare condition, and the causative mutation in Patient 2 was detected by NGS analysis; however, our flowchart also led to a diagnosis of HSH in this patient.
Loss-of-function mutations in the genes for claudin-16 and its close relative claudin-19 lead to an identical renal phenotype, with combined renal Ca2+ and Mg2+ wasting and nephrocalcinosis, referred to as FHHNC, inherited in an autosomal dominant mode [23, 24]. FHHNC is frequently complicated by progressive renal failure and the renal prognosis is poor, with progressive chronic kidney disease requiring renal replacement therapy typically occurring in the second or third decades of life [35]. Patients with CLDN16 and CLDN19 variants may have different renal prognoses; CLDN19 mutations are associated with a higher risk of chronic kidney disease and end-stage renal disease, and ocular impairment occurs exclusively in patients with CLDN19 mutations [22]. Definitive genetic diagnosis may be useful to prediction of prognosis or phenotype in FHHNC patients.
Gain-of-function mutations in CASR cause ADH1, which is associated with hypocalcemia, relative hypercalciuria, and inadequate PTH secretion, and occasionally with hypomagnesemia [17]. In some cases, ADH1 is accompanied by hypokalemia, and this combination may be classified as type V Batter syndrome [18, 19]. In addition to HSH, ADH1 may also be misdiagnosed as primary hypoparathyroidism, as in Patient 4.
Loss-of-function mutations in SLC12A3 cause Gitelman syndrome, which is an autosomal recessive renal tubulopathy characterized by hypokalemic metabolic alkalosis with hypocalciuria and hypomagnesemia [28]. Type III Batter syndrome, which is caused by CLCNKB variants, frequently shows phenotypic overlap with Gitelman syndrome [25–27], and some type III Batter syndrome patients may show clinical features of Gitelman syndrome, such as hypomagnesemia and hypocalciuria [25, 26]. It is not possible to make a definite diagnosis in such cases without genetic testing; however, a definitive genetic diagnosis will allow better management of patients and appropriate pathophysiological investigation of the disease.
Our flowchart was useful in the five index cases of inherited hypomagnesemia, and was also validated in the previous cases of inherited hypomagnesemia. However, many genes are involved in inherited hypomagnesemia, some of which have been reported in too few cases to define their characteristics. For instance, hypomagnesemia associated with variants in KCNA1 [9] or loss-of-function in the EGFR gene [20] has only been reported in one pedigree and one patient, respectively, though there have been some reports of cases with hypomagnesemia treated with cetuximab, a monoclonal antibody directed against EGFR [36]. Phenotypic overlap, such as that between Gitelman syndrome and type III Bartter syndrome [25, 26], or diversity of clinical phenotypes associated with gain-of-function mutations in CASR causing ADH1 and type V Bartter syndrome, will complicate the flowchart [18, 19, 37]. Further studies of inherited hypomagnesemia are, therefore, required to improve the flowchart.
In conclusion, hereditary hypomagnesemia may be difficult to diagnose accurately because of its rarity and the variety of causative genes. Our flowchart for identifying responsible genes for hypomagnesemia provides a useful diagnostic tool, but some kinds of hereditary hypomagnesemia still require genetic testing to reach a definite diagnosis. NGS analysis will help to resolve clinical difficulties and improve the chance of making a definite diagnosis in patients with hereditary hypomagnesemia.
References
de Baaij JH, Hoenderop JG, Bindels RJ. Magnesium in man: implications for health and disease. Physiol Rev. 2015;95(1):1–46.
Viering DH, de Baaij JH, Walsh SB, Kleta R, Bockenhauer D. Genetic causes of hypomagnesemia, a clinical overview. Pediatr Nephrol. 2016. doi:10.1007/s00467-016-3416-3.
Pham PC, Pham PA, Pham SV, Pham PT, Pham PM, Pham PT. Hypomagnesemia: a clinical perspective. Int J Nephrol Renovasc Dis. 2014;7:219–30.
Kamiyoshi N, Nozu K, Urahama Y, Matsunoshita N, Yamamura T, Minamikawa S, Ninchoji T, Morisada N, Nakanishi K, Kaito H, et al. Pathogenesis of hypokalemia in autosomal dominant hypocalcemia type 1. Clin Exp Nephrol. 2016;20(2):253–7.
Adalat S, Woolf AS, Johnstone KA, Wirsing A, Harries LW, Long DA, Hennekam RC, Ledermann SE, Rees L, van’t Hoff W, et al. HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol. 2009;20(5):1123–31.
Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, Lindner T, Yamagata K, Ogata M, Tomonaga O, et al. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet. 1997;17(4):384–5.
Vivante A, Kohl S, Hwang DY, Dworschak GC, Hildebrandt F. Single-gene causes of congenital anomalies of the kidney and urinary tract (CAKUT) in humans. Pediatr Nephrol. 2014;29(4):695–704.
Meij IC, Koenderink JB, van Bokhoven H, Assink KF, Groenestege WT, de Pont JJ, Bindels RJ, Monnens LA, van den Heuvel LP, Knoers NV. Dominant isolated renal magnesium loss is caused by misrouting of the Na(+),K(+)-ATPase gamma-subunit. Nat Genet. 2000;26(3):265–6.
Glaudemans B, van der Wijst J, Scola RH, Lorenzoni PJ, Heister A, van der Kemp AW, Knoers NV, Hoenderop JG, Bindels RJ. A missense mutation in the Kv1.1 voltage-gated potassium channel-encoding gene KCNA1 is linked to human autosomal dominant hypomagnesemia. J Clin Invest. 2009;119(4):936–42.
Stuiver M, Lainez S, Will C, Terryn S, Gunzel D, Debaix H, Sommer K, Kopplin K, Thumfart J, Kampik NB, et al. CNNM2, encoding a basolateral protein required for renal Mg2 + handling, is mutated in dominant hypomagnesemia. Am J Hum Genet. 2011;88(3):333–43.
Ferre S, de Baaij JH, Ferreira P, Germann R, de Klerk JB, Lavrijsen M, van Zeeland F, Venselaar H, Kluijtmans LA, Hoenderop JG, et al. Mutations in PCBD1 cause hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol. 2014;25(3):574–86.
de Baaij JH, Dorresteijn EM, Hennekam EA, Kamsteeg EJ, Meijer R, Dahan K, Muller M, van den Dorpel MA, Bindels RJ, Hoenderop JG, et al. Recurrent FXYD2 p.Gly41Arg mutation in patients with isolated dominant hypomagnesaemia. Nephrol Dial Transplant. 2015;30(6):952–7.
Astor MC, Lovas K, Wolff AS, Nedrebo B, Bratland E, Steen-Johnsen J, Husebye ES. Hypomagnesemia and functional hypoparathyroidism due to novel mutations in the Mg-channel TRPM6. Endocr Connect. 2015;4(4):215–22.
Groenestege WM, Hoenderop JG, van den Heuvel L, Knoers N, Bindels RJ. The epithelial Mg2+ channel transient receptor potential melastatin 6 is regulated by dietary Mg2+ content and estrogens. J Am Soc Nephrol. 2006;17(4):1035–43.
Schlingmann KP, Weber S, Peters M, Niemann Nejsum L, Vitzthum H, Klingel K, Kratz M, Haddad E, Ristoff E, Dinour D, et al. Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat Genet. 2002;31(2):166–70.
Unger S, Gorna MW, Le Bechec A, Do Vale-Pereira S, Bedeschi MF, Geiberger S, Grigelioniene G, Horemuzova E, Lalatta F, Lausch E, et al. FAM111A mutations result in hypoparathyroidism and impaired skeletal development. Am J Hum Genet. 2013;92(6):990–5.
Pearce SH, Williamson C, Kifor O, Bai M, Coulthard MG, Davies M, Lewis-Barned N, McCredie D, Powell H, Kendall-Taylor P, et al. A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med. 1996;335(15):1115–22.
Vargas-Poussou R, Huang C, Hulin P, Houillier P, Jeunemaitre X, Paillard M, Planelles G, Dechaux M, Miller RT, Antignac C. Functional characterization of a calcium-sensing receptor mutation in severe autosomal dominant hypocalcemia with a Bartter-like syndrome. J Am Soc Nephrol. 2002;13(9):2259–66.
Watanabe S, Fukumoto S, Chang H, Takeuchi Y, Hasegawa Y, Okazaki R, Chikatsu N, Fujita T. Association between activating mutations of calcium-sensing receptor and Bartter’s syndrome. The Lancet. 2002;360(9334):692–4.
Campbell P, Morton PE, Takeichi T, Salam A, Roberts N, Proudfoot LE, Mellerio JE, Aminu K, Wellington C, Patil SN, et al. Epithelial inflammation resulting from an inherited loss-of-function mutation in EGFR. J Invest Dermatol. 2014;134(10):2570–8.
Thebault S, Alexander RT, Tiel Groenestege WM, Hoenderop JG, Bindels RJ. EGF increases TRPM6 activity and surface expression. J Am Soc Nephrol. 2009;20(1):78–85.
Godron A, Harambat J, Boccio V, Mensire A, May A, Rigothier C, Couzi L, Barrou B, Godin M, Chauveau D, et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: phenotype-genotype correlation and outcome in 32 patients with CLDN16 or CLDN19 mutations. Clin J Am Soc Nephrol. 2012;7(5):801–9.
Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, Vitzthum H, Suzuki Y, Luk JM, Becker C, et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet. 2006;79(5):949–57.
Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, Casari G, Bettinelli A, Colussi G, Rodriguez-Soriano J, et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2 + resorption. Science. 1999;285(5424):103–6.
Matsunoshita N, Nozu K, Shono A, Nozu Y, Fu XJ, Morisada N, Kamiyoshi N, Ohtsubo H, Ninchoji T, Minamikawa S, et al. Differential diagnosis of Bartter syndrome, Gitelman syndrome, and pseudo-Bartter/Gitelman syndrome based on clinical characteristics. Genet Med. 2016;18(2):180–8.
Nozu K, Iijima K, Kanda K, Nakanishi K, Yoshikawa N, Satomura K, Kaito H, Hashimura Y, Ninchoji T, Komatsu H, et al. The pharmacological characteristics of molecular-based inherited salt-losing tubulopathies. J Clin Endocrinol Metab. 2010;95(12):E511–E518.
Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, Schurman S, Nayir A, Alpay H, Bakkaloglu A, et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet. 1997;17(2):171–8.
Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, et al. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na–Cl cotransporter. Nat Genet. 1996;12(1):24–30.
Birkenhager R, Otto E, Schurmann MJ, Vollmer M, Ruf EM, Maier-Lutz I, Beekmann F, Fekete A, Omran H, Feldmann D, et al. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet. 2001;29(3):310–4.
Scholl UI, Choi M, Liu T, Ramaekers VT, Hausler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci USA. 2009;106(14):5842–7.
Cruz DN, Shaer AJ, Bia MJ, Lifton RP, Simon DB, Yale Gs, Bartter’s Syndrome Collaborative Study G. Gitelman’s syndrome revisited: an evaluation of symptoms and health-related quality of life. Kidney Int. 2001;59(2):710–7.
Lainez S, Schlingmann KP, van der Wijst J, Dworniczak B, van Zeeland F, Konrad M, Bindels RJ, Hoenderop JG. New TRPM6 missense mutations linked to hypomagnesemia with secondary hypocalcemia. Eur J Hum Genet. 2014;22(4):497–504.
Mache CJ, Preisegger KH, Kopp S, Ratschek M, Ring E. De novo HNF-1 beta gene mutation in familial hypoplastic glomerulocystic kidney disease. Pediatr Nephrol. 2002;17(12):1021–6.
Watanabe T, Bai M, Lane CR, Matsumoto S, Minamitani K, Minagawa M, Niimi H, Brown EM, Yasuda T. Familial hypoparathyroidism: identification of a novel gain of function mutation in transmembrane domain 5 of the calcium-sensing receptor. J Clin Endocrinol Metab. 1998;83(7):2497–502.
Weber S, Schneider L, Peters M, Misselwitz J, Ronnefarth G, Boswald M, Bonzel KE, Seeman T, Sulakova T, Kuwertz-Broking E, et al. Novel paracellin-1 mutations in 25 families with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol. 2001;12(9):1872–81.
Schrag D, Chung KY, Flombaum C, Saltz L. Cetuximab therapy and symptomatic hypomagnesemia. J Natl Cancer Inst. 2005;97(16):1221–4.
Kinoshita Y, Hori M, Taguchi M, Watanabe S, Fukumoto S. Functional activities of mutant calcium-sensing receptors determine clinical presentations in patients with autosomal dominant hypocalcemia. J Clin Endocrinol Metab. 2014;99(2):E363–E368.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All procedures were approved by the Institutional Review Board (IRB) of Kobe University Graduate School of Medicine and in accordance with the Helsinki Declaration of 1975, as revised in 2000 (IRB number: 301). Informed consent was obtained from all index patients or their parents.
This study was supported by a grant from the Ministry of Education, Culture, Sports, Science and Technology of Japan (Subject ID: 15K09691 to Kandai Nozu) and a Health Labour Sciences Research Grant for Research on Measures for Intractable Diseases (H26-nanchitou-ippan-036 to Kazumoto Iijima).
Conflict of interest
The authors have nothing to disclose.
About this article

Cite this article
Horinouchi, T., Nozu, K., Kamiyoshi, N. et al. Diagnostic strategy for inherited hypomagnesemia. Clin Exp Nephrol 21, 1003–1010 (2017). https://doi.org/10.1007/s10157-017-1396-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10157-017-1396-7