
Abstract
Ischemia–reperfusion injury is a main cause of acute kidney injury. Tubular necrosis and interstitial inflammatory cell infiltration are characteristic pathologic changes of acute kidney injury. The main necrotic area should be repaired with new tubular epithelial cells after the injury. On the other hand, some parts of the injured kidney progress to interstitial fibrosis, a characteristic pathologic change in chronic kidney disease. We hypothesized that interstitial infiltrating leukocytes, that are attracted and activated by chemokines, are key mediators in the pathogenesis of tubular necrosis, regeneration of the necrotic area, or interstitial fibrosis. A large number of chemokines were upregulated after ischemic injury, and chemokine receptor-expressing inflammatory cells were attracted by these chemokines. Genetic or molecular modulating experiments in the mouse model have begun to reveal the key participants and their specific roles at the levels of inflammation, regeneration, and fibrosis. Among these chemokines/chemokine receptors, our data indicated CCR2-mediated macrophage infiltration mainly affected tubular necrosis after ischemic acute kidney injury, interferon-gamma-inducible protein (IP)-10-producing macrophages participate in regeneration of tubular epithelial cells, and CX3CR1-mediated macrophages and platelet infiltration and aggregation play roles in interstitial fibrosis in chronic kidney disease. These chemokines and chemokine receptors on infiltrating inflammatory cells would be novel clinical markers or targets for therapeutic intervention.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ischemia–reperfusion injury (IR) is one of main clinical causes of acute kidney injury. Ischemia–reperfusion is unavoidable during renal transplantation, and is also commonly observed in renal artery stenosis, and shock state occurring for a variety of reasons. From the viewpoint of pathogenesis of ischemic acute kidney injury, tubular necrosis and interstitial inflammatory cell infiltration are characteristic pathologic changes. Moreover, the necrotic area is repaired by new tubular epithelial cells several days after the injury. On the other hand, some parts of the injured kidney progress to interstitial fibrosis, a characteristic pathologic change of chronic kidney disease (Fig. 1). Inflammatory mechanisms resulting from ischemic acute kidney injury and leading to chronic kidney disease have not yet been fully defined but may provide opportunities for developing novel therapeutic approaches [1–3]. As well as therapeutic targets, to understand the precise mechanisms of progression to acute kidney injury with necrosis, regeneration of necrotic tubular epithelial cells or chronic kidney disease with interstitial fibrosis are important for developing new diagnostic markers. This review focuses on the contribution of inflammation regulated by cytokines and chemokines as a key modulator in renal ischemia–reperfusion injury.

Natural course of ischemia–reperfusion injury. In acute kidney injury after ischemia, tubular necrosis and interstitial inflammatory cell infiltration are observed in the early phase of injury. The necrotic area is repaired by new tubular epithelial cells. Interstitial fibrosis, a characteristic pathologic change of chronic kidney disease, progresses several days after the injury
Tubular necrosis
Induction of chemokines after renal ischemia–reperfusion injury
Inflammatory cell infiltration in ischemic acute kidney injury might be one of key responses in the progression of tissue destruction and regeneration of the injured kidney. At least three mechanisms of action induce inflammatory responses:
-
The first is adenosine triphosphate (ATP) depression. Oxygen deprivation due to ischemia induces early ATP depletion. ATP depletion stops ATP-dependent transport pumps, resulting in mitochondrial swelling. Mitochondrial swelling results in outer membrane rupture, with release of mitochondrial intermembrane proteins. Caspase 1 or interleukin-1 converting enzyme (ICE) enzymatically cleaves interleukin (IL)-1β. IL-1β is an exponent pro-inflammatory cytokine, and can induce chemokines, for example keratinocyte-induced chemoattractant (KC), macrophage inflammatory protein (MIP)-1α, or RANTES, from renal tubular epithelial cells [4].
-
A second species mediating ischemia-induced inflammation in the kidney is hypoxia inducible factor-1 (HIF-1). Three isoforms of HIF have been identified, HIF-1, HIF-2, and HIF-3. HIF-1 and HIF-2 are structurally and functionally similar. However, HIF-3 may be a negative regulator of hypoxia-inducible genes expressed by HIF-1 and HIF-2 [5]. HIF-1 is unstable under normoxic conditions but is stable and works under hypoxic conditions [6, 7]. Many genes encoded for cytokines and growth factors are induced by HIF-1 activation [8, 9].
-
The third molecular switch linking ischemia to inflammation involves oxygen-derived free radicals. In particular, hydrogen peroxide, which is a source of oxygen-derived free radicals after IR injury, has been reported to induce TNF-α production by activating p38 mitogen-activated protein kinase (MAPK) [10].
These mechanisms combine to induce inflammation after ischemia–reperfusion, and subsequent mechanisms augment the inflammation and tubular necrosis.
Intracellular signaling pathways linking ischemic injury to chemokine production
A variety of intracellular signaling factors, for example nuclear factor (NF)-κB and activating protein (AP)-1, are necessary for CXCL8/interleukin (IL)-8, CCL2/monocyte chemoattractant protein (MCP)-1, or CCL5/regulated upon activation normal T cells expressed and secreted (RANTES) [11–13]. Activation of NF-κB and AP-1 requires phosphorylation of p38 MAPK. Consistent with this, we found that pharmacological inhibition of p38 MAPK significantly reduced inflammatory cytokine and chemokine production and prevented tubular necrosis in a mouse model of renal IR injury [4]. In the same way, NF-κB decoy oligodeoxynucleotide treatment attenuated tubular necrosis with reduction of CCL2/MCP-1 expression in a rat model of renal IR injury [14]. Thus, these intracellular signaling factors may be good targets for therapeutic intervention in renal IR injury. However, further studies will be needed to clarify the importance and significance of each intracellular signaling cascade on cytokine and chemokine expression in renal IR injury.
Pro-inflammatory cytokines augment inflammation after renal ischemia–reperfusion injury
Renal parenchymal cells, for example tubular epithelial cells, mesangial cells, and endothelial cells, have the potential to produce various kinds of chemokine (e.g. CCL2/MCP-1, CCL5/RANTES, CXCL8/IL-8, CXCL1/growth-regulated oncogene (GRO), CXCL10/interferon-gamma inducible protein (IP)-10, CXCL2/3/MIP-2, etc.) in response to stimulation with pro-inflammatory cytokines such as IL-1β, TNF-α, and interferon (IFN)-γ, immune complexes, and growth factors, including platelet-derived growth factor and basic fibroblast growth factor [15]. Midkine also enhances migration of inflammatory cells on ischemic injury of the kidney, and may induce chemokine production and ischemic tissue damage [16, 17]. Among chemokine/cytokine-producing cells in the kidney, the tubular epithelial cells seem to be the most sensitive to hypoxic conditions; moreover, tubular cell-derived cytokines/chemokines are thought to augment inflammatory processes in renal ischemia–reperfusion injury.
Chemokine expression in the injured kidney
Chemokines are able to activate many different cell types, including leukocytes and renal parenchymal cells. They have been divided into four subfamilies—CXC, CC, C, and CX3C—according to the number and spacing of conserved cysteine residues in their sequences.
A large number of chemokines and chemokine receptors are expressed after ischemia–reperfusion injury. The CXC chemokines, for example KC and CXCL2/3/MIP-2 in the mouse and CXCL8/IL-8 in man, rabbit, and other mammals, are upregulated in tubular epithelial cells after ischemic injury; this results in marked neutrophil infiltration [18–22]. CXCR2, a receptor for KC and CXCL2/3MIP-2 and CXCL8/IL-8, is one therapeutic target for neutrophil infiltration in a mouse model of renal IR injury [20, 23–25]. However, neutrophil-depletion studies with anti-neutrophil antibodies in mouse models have not provided evidence of an important role of neutrophils in IR [26]. CC chemokines are also upregulated after ischemic injury, with marked monocyte/macrophage infiltration. In contrast with neutrophil depletion, macrophage depletion with clodronate in vivo has furnished evidence of a major role in IR pathogenesis of monocytes/macrophages, which accumulate in the late phase post-injury [27]. Expression of the monocyte-targeted CC chemokine CCL2/MCP-1 is induced in ischemic kidney [28]. Both genetic deletion and pharmacological blockade of CCR2, the specific receptor for CCL2/MCP-1, have been reported to significantly reduce monocyte infiltration and tubular necrosis after ischemic injury [4, 29, 30]. Immunohistological studies indicate that the main resident cell producing CCL2/MCP-1 in the kidney after ischemic injury is located in the distal tubule [31]. Macrophages are also a major source of CCL2/MCP-1, and of the inflammatory cytokines TNF-α, IL-1, and IL-6 [32]. Therefore, these MCP-1/CCR2 positive-feedback loops augment inflammatory reaction after ischemia–reperfusion injury and might participate in progression of tubular necrosis.
Regeneration
Acute kidney injury with tubular necrosis usually recovers after the injury. During the recovery phase, necrotic tubular cell debris is removed and the denuded area is restored by new tubular cells [33]. Although tubular cell regeneration is undoubtedly required for recovery of renal function, the precise mechanisms and factors regulating tubular cell proliferation are poorly understood [34]. From the viewpoint of sources of regenerating tubular cells, several kinds of stem cell have been speculated upon. Bone marrow-derived stem cells or intra-renal mesenchymal stem cells might participate in the regeneration [35], but residual tubular epithelial cells aggressively proliferate to cover the area of the defect. Tubular epithelial cells may produce growth factors (FGF, TGF-α, EGF-like growth factor, IL-2, osteopontin, etc.) at regenerative wound margins associated with leukocyte infiltration [36–38]. It is likely that inflammatory infiltrating leukocytes secrete cytokines and chemokines that may regulate growth-factor production by resident renal cells. Concurrent with growth factor production, regenerative changes of cell proliferation associated with leukocyte infiltration have been detected at wound margins. However, excessive cell proliferation should be harmful for perfect recovery of the injured kidney. Therefore, as well as cell proliferation, inhibitory mechanisms of cell proliferation might be necessary for perfect regeneration. On this topic we reported that IP-10 had an inhibitory effect on tubular cell proliferation in vivo and in vitro. The interstitial infiltrated F4/80-positive cells were main source of IP-10 [39]. Further studies are needed to investigate whether regeneration from tubular necrosis is a very important part of recovery of the injured kidney.
Interstitial fibrosis after renal ischemia–reperfusion injury
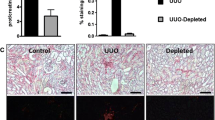
The main parts of ischemic acute kidney injury regenerate and recover renal function. However, some parts progress to interstitial fibrosis, which is one of the characteristic changes in chronic kidney disease and an important poor prognostic factor. It is well known that the fibroblast is a key fibrogenic cell, but we recently reported the importance of circulating connective tissue cell progenitors, fibrocytes, to interstitial fibrosis [40]. In parallel with fibrogenic cells, humoral factors are also important in the progression of kidney fibrosis. Transforming growth factor (TGF)-β and platelet-derived growth factor (PDGF) are well-characterized factors that promote fibrosis in many diseases and organs, including the kidney [41, 42]. As well as tubular epithelial cells and fibroblasts, infiltrating inflammatory cells are rich sources of these factors and, potentially, other fibrogenic mediators [43]. We revealed that CX3CL1/fractalkine and its unique receptor CX3CR1 are both upregulated after ischemic injury and participate in the pathogenesis of renal fibrosis. The mechanism involves selective effects in the outer medulla, including accumulation of macrophages and platelets, and expression of the macrophage and platelet-derived fibrogenic protein platelet-derived growth factor-B [44].
Anti-inflammatory treatment approaches in renal ischemia–reperfusion injury
Prognosis of acute kidney injury has not changed in the past two decades. New approaches to the disease are necessary for prevention of poor prognosis. We showed the inflammatory reaction might be a key aspect of progression of the disease. Although vasoactive agents such as dopamine, atrial natriuretic peptide, and diuretics are clinically useful for acute kidney injury after renal IR injury, these agents do not directly affect inflammation. Several experimental therapeutic approaches, for example leukocyte depletion, and anti-cytokine, anti-chemokine, and anti-adhesion molecule therapies, have been tested with the objective of controlling renal inflammation after IR injury.
Inhibition of caspase activation is one target for therapeutic treatment of ischemic acute kidney injury. Treatment with the caspase inhibitor Z–Val–Ala–Asp(OMe)-CH2F effectively diminishes apoptosis, chemokine expression, and neutrophil infiltration after renal ischemia–reperfusion [22].
Intracellular signaling molecules are also therapeutic targets for ischemic acute kidney injury. Inhibitors of NF-κB reduce CCL2/MCP-1 mRNA expression and prevent tissue destruction in ischemic acute kidney injury [28]. We targeted p38 MAPK, a key factor regulating inflammatory chemokine expression, for treatment of ischemic renal injury. An inhibitor of p38MAPK was effective in significantly reducing chemokine expression and in attenuating leukocyte infiltration and tissue destruction [4].
Chemokines and chemokine receptors themselves are potentially important targets of anti-inflammatory therapy. We previously reported that neutralizing anti-IL-8 antibodies or anti-MCP-1 antibodies prevents experimental glomerulonephritis [45, 46]. Moreover, specific inhibitors of chemokine receptors, for example vMIP-II, APO-RANTES or Met-RANTES, 7ND MCP-1, TAK-779, or propagermanium, have been developed and tested successfully in various animal models, including experimental renal IR injury [29, 30, 47–51]. Furthermore, we revealed that neutralizing anti-CX3CR1 antibodies prevents interstitial fibrosis with diminishing macrophage infiltration [44]. These reports are encouraging, and add support to consideration of inflammatory chemokine receptors as targets for IR injury in the clinic.
Conclusion
Interstitial infiltrating leukocytes are key mediators in the pathogenesis of acute kidney injury with tubular necrosis, in the regeneration of necrotic tubular epithelial cells, and in chronic kidney disease with interstitial fibrosis. Various kinds of inflammatory infiltrating cell are mediated by cytokines and chemokines. Many chemokines are upregulated after ischemic injury, and chemokine receptor-expressing inflammatory cells are attracted by these chemokines. Genetic or molecular-modulating experiments in the mouse model have begun to reveal the key players and their specific roles at the levels of inflammation, regeneration, and fibrosis. Among these chemokines/chemokine receptors, our data indicated CCR2-mediated macrophage infiltration mainly affected tubular necrosis after ischemic acute kidney injury, IP-10-producing macrophages participate in the regeneration of tubular epithelial cells, and CX3CR1-mediated macrophages and platelets participate in interstitial fibrosis in chronic kidney disease (Fig. 2). Other major challenges will be to expand these approaches and to translate emerging results for use in human diseases in which renal IR injury occurs, with the objectives of identifying novel clinical markers or targets for therapeutic intervention.

Chemokine cascades in ischemia–reperfusion injury pathologic change of tubular necrosis after ischemic acute kidney injury are mainly mediated by the action of CCR2 on macrophages. Regeneration of tubular epithelial cells in the late phase of the injury was mediated by IP-10-producing macrophages. Moreover, chronic interstitial fibrosis was mediated by the action of CX3CR1 on macrophages and platelets. Characteristic chemokines/chemokine receptors participate in particular pathologic changes at specific time points
References
Frangogiannis NG. Chemokines in ischemia and reperfusion. Thromb Haemost. 2007;97:738–47.
Minami M, Satoh M. Chemokines and their receptors in the brain: pathophysiological roles in ischemic brain injury. Life Sci. 2003;74:321–7.
Frangogiannis NG, Entman ML. Chemokines in myocardial ischemia. Trends Cardiovasc Med. 2005;15:163–9.
Furuichi K, Wada T, Iwata Y, Sakai N, Yoshimoto K, Kobayashi Ki K, et al. Administration of FR167653, a new anti-inflammatory compound, prevents renal ischaemia/reperfusion injury in mice. Nephrol Dial Transplant. 2002;17:399–407.
Nangaku M, Inagi R, Miyata T, Fujita T. Hypoxia and hypoxia-inducible factor in renal disease. Nephron Exp Nephrol. 2008;110:e1–7.
Huang LE, Arany Z, Livingston DM, Bunn HF. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J Biol Chem. 1996;271:32253–9.
Salceda S, Caro J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem. 1997;272:22642–7.
El Awad B, Kreft B, Wolber EM, Hellwig-Burgel T, Metzen E, Fandrey J, et al. Hypoxia and interleukin-1beta stimulate vascular endothelial growth factor production in human proximal tubular cells. Kidney Int. 2000;58:43–50.
Zhou J, Brune B. Cytokines and hormones in the regulation of hypoxia inducible factor-1alpha (HIF-1alpha). Cardiovasc Hematol Agents Med Chem. 2006;4:189–97.
Meldrum DR, Dinarello CA, Cleveland JC Jr, Cain BS, Shames BD, Meng X, et al. Hydrogen peroxide induces tumor necrosis factor alpha-mediated cardiac injury by a P38 mitogen-activated protein kinase-dependent mechanism. Surgery. 1998;124:291–6; discussion 297.
Nelson PJ, Ortiz BD, Pattison JM, Krensky AM. Identification of a novel regulatory region critical for expression of the RANTES chemokine in activated T lymphocytes. J Immunol. 1996;157:1139–48.
Yasumoto K, Okamoto S, Mukaida N, Murakami S, Mai M, Matsushima K. Tumor necrosis factor alpha and interferon gamma synergistically induce interleukin 8 production in a human gastric cancer cell line through acting concurrently on AP-1 and NF-kB-like binding sites of the interleukin 8 gene. J Biol Chem. 1992;267:22506–11.
Lenardo MJ, Baltimore D. NF-kappa B: a pleiotropic mediator of inducible and tissue-specific gene control. Cell. 1989;58:227–9.
Cao CC, Ding XQ, Ou ZL, Liu CF, Li P, Wang L, et al. In vivo transfection of NF-kappaB decoy oligodeoxynucleotides attenuate renal ischemia/reperfusion injury in rats. Kidney Int. 2004;65:834–45.
Segerer S, Nelson PJ, Schlondorff D. Chemokines, chemokine receptors, and renal disease: from basic science to pathophysiologic and therapeutic studies. J Am Soc Nephrol. 2000;11:152–76.
Sato W, Kadomatsu K, Yuzawa Y, Muramatsu H, Hotta N, Matsuo S, et al. Midkine is involved in neutrophil infiltration into the tubulointerstitium in ischemic renal injury. J Immunol. 2001;167:3463–9.
Sato W, Takei Y, Yuzawa Y, Matsuo S, Kadomatsu K, Muramatsu T. Midkine antisense oligodeoxyribonucleotide inhibits renal damage induced by ischemic reperfusion. Kidney Int. 2005;67:1330–9.
Thurman JM, Lenderink AM, Royer PA, Coleman KE, Zhou J, Lambris JD, et al. C3a is required for the production of CXC chemokines by tubular epithelial cells after renal ischemia/reperfusion. J Immunol. 2007;178:1819–28.
Molls RR, Savransky V, Liu M, Bevans S, Mehta T, Tuder RM, et al. Keratinocyte-derived chemokine is an early biomarker of ischemic acute kidney injury. Am J Physiol Renal Physiol. 2006;290:F1187–93.
Cugini D, Azzollini N, Gagliardini E, Cassis P, Bertini R, Colotta F, et al. Inhibition of the chemokine receptor CXCR2 prevents kidney graft function deterioration due to ischemia–reperfusion. Kidney Int. 2005;67:1753–61.
Miura M, Fu X, Zhang QW, Remick DG, Fairchild RL. Neutralization of Gro alpha and macrophage inflammatory protein-2 attenuates renal ischemia/reperfusion injury. Am J Pathol. 2001;159:2137–45.
Daemen MA, de Vries B, van’t Veer C, Wolfs TG, Buurman WA. Apoptosis and chemokine induction after renal ischemia/reperfusion. Transplantation. 2001;71:1007–11.
Popivanova BK, Koike K, Tonchev AB, Ishida Y, Kondo T, Ogawa S, et al. Accumulation of microglial cells expressing ELR motif-positive CXC chemokines and their receptor CXCR2 in monkey hippocampus after ischemia–reperfusion. Brain Res. 2003;970:195–204.
Tarzami ST, Miao W, Mani K, Lopez L, Factor SM, Berman JW, et al. Opposing effects mediated by the chemokine receptor CXCR2 on myocardial ischemia–reperfusion injury: recruitment of potentially damaging neutrophils and direct myocardial protection. Circulation. 2003;108:2387–92.
Belperio JA, Keane MP, Burdick MD, Gomperts BN, Xue YY, Hong K, et al. CXCR2/CXCR2 ligand biology during lung transplant ischemia–reperfusion injury. J Immunol. 2005;175:6931–9.
Melnikov VY, Faubel S, Siegmund B, Lucia MS, Ljubanovic D, Edelstein CL. Neutrophil-independent mechanisms of caspase-1- and IL-18-mediated ischemic acute tubular necrosis in mice. J Clin Invest. 2002;110:1083–91.
Day YJ, Huang L, Ye H, Linden J, Okusa MD. Renal ischemia–reperfusion injury and adenosine 2A receptor-mediated tissue protection: role of macrophages. Am J Physiol Renal Physiol. 2005;288:F722–31.
Sung FL, Zhu TY, Au-Yeung KK, Siow YL, Karmin O. Enhanced MCP-1 expression during ischemia/reperfusion injury is mediated by oxidative stress and NF-kappaB. Kidney Int. 2002;62:1160–70.
Furuichi K, Wada T, Iwata Y, Kitagawa K, Kobayashi K, Hashimoto H, et al. Gene therapy expressing amino-terminal truncated monocyte chemoattractant protein-1 prevents renal ischemia–reperfusion injury. J Am Soc Nephrol. 2003;14:1066–71.
Furuichi K, Wada T, Iwata Y, Kitagawa K, Kobayashi K, Hashimoto H, et al. CCR2 signaling contributes to ischemia–reperfusion injury in kidney. J Am Soc Nephrol. 2003;14:2503–15.
Rice JC, Spence JS, Yetman DL, Safirstein RL. Monocyte chemoattractant protein-1 expression correlates with monocyte infiltration in the post-ischemic kidney. Ren Fail. 2002;24:703–23.
Jo SK, Sung SA, Cho WY, Go KJ, Kim HK. Macrophages contribute to the initiation of ischaemic acute renal failure in rats. Nephrol Dial Transplant. 2006;21:1231–9.
Liu KD. Molecular mechanisms of recovery from acute renal failure. Crit Care Med. 2003;31:S572–81.
Nony PA, Schnellmann RG. Mechanisms of renal cell repair and regeneration after acute renal failure. J Pharmacol Exp Ther. 2003;304:905–12.
Bussolati B, Tetta C, Camussi G. Contribution of stem cells to kidney repair. Am J Nephrol. 2008;28:813–22.
Ghielli M, Verstrepen W, Nouwen E, De Broe ME. Regeneration processes in the kidney after acute injury: role of infiltrating cells. Exp Nephrol. 1998;6:502–7.
Naor D, Sionov RV, Ish-Shalom D. CD44: structure, function, and association with the malignant process. Adv Cancer Res. 1997;71:241–319.
Lewington AJ, Padanilam BJ, Martin DR, Hammerman MR. Expression of CD44 in kidney after acute ischemic injury in rats. Am J Physiol Regul Integr Comp Physiol. 2000;278:R247–54.
Furuichi K, Wada T, Kitajikma S, Toyama T, Okumura T, Hara A, et al. IFN-inducible protein 10 (CXCL10) regulates tubular cell proliferation in renal ischemia–reperfusion injury. Nephron Exp Nephrol. 2008;109:c29–38.
Wada T, Sakai N, Matsushima K, Kaneko S. Fibrocytes: a new insight into kidney fibrosis. Kidney Int. 2007;72:269–73.
Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med. 1994;331:1286–92.
Hugo C. The thrombospondin 1-TGF-beta axis in fibrotic renal disease. Nephrol Dial Transplant. 2003;18:1241–5.
Hirschberg R, Wang S. Proteinuria and growth factors in the development of tubulointerstitial injury and scarring in kidney disease. Curr Opin Nephrol Hypertens. 2005;14:43–52.
Furuichi K, Gao JL, Murphy PM. Chemokine receptor CX3CR1 regulates renal interstitial fibrosis after ischemia–reperfusion injury. Am J Pathol. 2006;169:372–87.
Wada T, Tomosugi N, Naito T, Yokoyama H, Kobayashi K, Harada A, et al. Prevention of proteinuria by the administration of anti-interleukin 8 antibody in experimental acute immune complex-induced glomerulonephritis. J Exp Med. 1994;180:1135–40.
Wada T, Yokoyama H, Furuichi K, Kobayashi KI, Harada K, Naruto M, et al. Intervention of crescentic glomerulonephritis by antibodies to monocyte chemotactic and activating factor (MCAF/MCP-1). FASEB J. 1996;10:1418–25.
Wada T, Yokoyama H, Matsushima K, Kobayashi K. Chemokines in renal diseases. Int Immunopharmacol. 2001;1:637–45.
Cascieri MA, Springer MS. The chemokine/chemokine-receptor family: potential and progress for therapeutic intervention. Curr Opin Chem Biol. 2000;4:420–7.
Proudfoot AE. Chemokine receptors: multifaceted therapeutic targets. Nat Rev Immunol. 2002;2:106–15.
Grone HJ, Weber C, Weber KS, Grone EF, Rabelink T, Klier CM, et al. Met-RANTES reduces vascular and tubular damage during acute renal transplant rejection: blocking monocyte arrest and recruitment. FASEB J. 1999;13:1371–83.
Kitagawa K, Wada T, Furuichi K, Hashimoto H, Ishiwata Y, Asano M, et al. Blockade of CCR2 ameliorates progressive fibrosis in kidney. Am J Pathol. 2004;165:237–46.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Furuichi, K., Kaneko, S. & Wada, T. Chemokine/chemokine receptor-mediated inflammation regulates pathologic changes from acute kidney injury to chronic kidney disease. Clin Exp Nephrol 13, 9–14 (2009). https://doi.org/10.1007/s10157-008-0119-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10157-008-0119-5