
Abstract
Aldosterone is traditionally viewed as a hormone regulating electrolyte and blood pressure homeostasis by acting on the distal nephron. Accumulating evidence suggests that aldosterone also plays pathogenetic roles in cardiovascular and renal injury. For example, aldosterone is a potent inducer of proteinuria. We demonstrated that podocyte injury underlies the pathogenesis of proteinuria in aldosterone-infused rats on a high salt diet. Mineralocorticoid receptor was detected in the podocytes in vivo and in vitro, and aldosterone caused induction of its effector kinase Sgk1, activation of NADPH oxidase and generation of reactive oxygen species. Selective aldosterone blocker eplerenone, as well as antioxidant tempol, ameliorated aldosterone-induced podocyte injury and proteinuria. Aldosterone was also involved in the podocyte damage and proteinuria of metabolic syndrome model SHR/NDmcr-cp. Adipocyte-derived aldosterone releasing factors were suggested to contribute to the aldosterone excess of this model. Furthermore, high salt diet markedly worsened the renal injury of SHR/NDmcr-cp. Although salt lowered serum aldosterone levels, it caused MR activation in the kidney. Accordingly, eplerenone dramatically improved the salt-evoked nephropathy. Taken together, aldosterone blockers can be an excellent therapeutic strategy for the treatment of podocyte injury, proteinuria, and cardiovascular and renal complications, not only in high aldosterone states but also in patients with activated MR signaling in the target tissue, whose circulating aldosterone level is not necessarily high. Addition of aldosterone blockers in patients treated with ACEIs or ARBs are also promising, because of “aldosterone breakthrough” phenomenon. Careful monitoring of hyperkalemia is necessary, especially in patients with impaired renal function.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since its isolation in 1953 [1], aldosterone (formerly called “electrocortin”) has been recognized as a hormone regulating electrolyte, volume, and blood pressure (BP) homeostasis [2]. This classic role of aldosterone was substantiated by many genetic disorders of aldosterone cascade in the distal nephron manifesting combinations of electrolyte, BP, and acid/base perturbations [3, 4]. Recently, however, growing evidence suggests that aldosterone plays a pathogenetic role in mediating cardiovascular injury [5–7]. Aldosterone has been shown to act on nonepithelial cells in the heart, vasculature, and brain to cause tissue remodeling, fibrosis, and endothelial dysfunction. This nonclassic mode of action was highlighted by the Randomized Aldactone Evaluation Study (RALES) and the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS), in which patients with congestive heart failure benefited from aldosterone blockers [8, 9]. Accumulating lines of evidence also indicate the role of aldosterone in progressive kidney disease [5, 10].
In this review, we summarize the recent advances in aldosterone research and introduce our data regarding aldosterone-induced podocyte damage and proteinuria and its underlying mechanisms [11, 12]. We also show the role of aldosterone in the nephropathy associated with metabolic syndrome, and the possible contribution of adipocyte-derived aldosterone-releasing factors [13, 14].
Classic actions of aldosterone
Aldosterone is traditionally viewed as a hormone controlling sodium reabsorption and potassium excretion in the distal nephron. The direct targets for aldosterone (aldosterone-sensitive distal nephron: ASDN) are the late distal convoluted tubules (DCT2), the connecting tubules (CNT), and the principal cells of the cortical and medullary collecting ducts (Table 1). In these cells, aldosterone activates the amiloride-sensitive epithelial sodium channel (ENaC), thiazide-sensitive sodium chloride cotransporter (NCC), and Na, K-ATPase [15–17]. These segments express not only mineralocorticoid receptor (MR) but also 11β hydroxysteroid dehydrogenase type 2, which catabolizes glucocorticoids into inactive metabolites, exposing these cells to mineralocorticoids. Aldosterone binds to MR, a member of the nuclear receptor superfamily, and translocates into the nucleus. The ligand-receptor complex then forms a homodimer, binds to the specific DNA sequences and stimulates the transcription of target genes, such as serum- and glucocorticoid-inducible kinase 1 (Sgk1), ENaC α subunit, and the glucocorticoid-induced leucine zipper (GILZ) protein. Sgk1, a serine-threonine kinase, is an important mediator of aldosterone action [18, 19]. Sgk1 is activated by phosphatidylinositol 3-kinase which then phosphorylates a ubiquitin protein ligase Nedd4-2 and reduces Nedd4-2-mediated proteasomal degradation of ENaC [7]. Sgk1 also stimulates various renal transport proteins including the renal outer medullary K channel ROMK (Kir1.1) and voltage gated potassium channel Kv1.3. In addition, recent evidence clarified that aldosterone modulates the functions of ENaC, NCC, and ROMK via With-No-Lysine [K] (WNK) 1 and 4, another family of serine-threonine kinases [20].
Gene targeting of ENaC, Skg1, as well as MR, resulted in salt-wasting in mice, especially when the animals were fed a low-sodium diet [21–25]. The findings confirm the physiological significance of these aldosterone effectors in sodium homeostasis.
Aldosterone as a mediator of target organ damage
Recently, there has been a paradigm shift regarding the role of aldosterone [26]. Growing evidence suggests that aldosterone plays important pathogenetic roles in cardiovascular and renal injury, and is postulated as a “cardiovascular risk hormone”. Aldosterone together with increased salt intake produces perivascular inflammation and fibrosis in the heart [27]. Aldosterone causes endothelial dysfunction and vascular remodeling. Some of these effects of aldosterone are reported to be mediated by rapid non-genomic actions [28]. In the kidney, aldosterone promotes proteinuria, glomerulosclerosis, arteriopathy, and renal fibrosis [29, 30]. MR is reported to be detected not only in the distal nephron but also in the glomerular mesangial cells, renal fibroblasts, and vascular smooth muscle cells [31–34]. Multiple factors are proposed to be involved: plasminogen activator inhibitor (PAI)-1, transforming growth factor (TGF)-β1, collagens I, III, IV, fibronectin, connective tissue growth factor, reactive oxygen species (ROS), proinflammatory cytokines such as osteopontin and monocyte chemoattractant protein (MCP)-1, and upregulation of angiotensin (Ang) II type 1 receptor [5, 10]. ROS are considered as important mediators, [30, 31, 35–37] which activate extracellular signal-regulated kinase 1/2, c-Jun N-terminal kinase, and big mitogen-activated protein kinase (BMK1) but not p38 mitogen-activated protein kinase in rat renal cortex and cultured mesangial cells [30, 38]. The small GTP-binding protein RhoA is another factor mediating aldosterone-induced renal injury. Sun et al. [39] demonstrated that fasudil, a specific Rho-kinase inhibitor, protected from proteinuria, glomerular cell proliferation, and tubulointerstitial fibrosis in uninephrectomized aldosterone/salt-loaded rats, although it did not change BP.
Aldosterone and proteinuria
Proteinuria is an important component in the definition of chronic kidney disease (CKD) by the Kidney Disease Outcome Quality Initiative (K/DOQI) [40]. Accumulating evidence suggests that aldosterone is a potent inducer of proteinuria [5, 10, 41, 42]. Clinical trials have proven the anti-proteinuric effects of angiotensin I-converting enzyme inhibitors (ACEIs) and angiotensin II (Ang II) type 1 receptor blockers (ARBs) in patients with diabetic and non-diabetic nephropathy, and Ang II is thought to be the primary responsible factor [43]. However, several lines of evidence implicate the role for aldosterone as well. For example, Greene et al. [44] demonstrated that proteinuria and renal injury in the real ablation rats were associated with elevated plasma aldosterone, and that anti-proteinuric effects of ACEI and ARB were abrogated by exogenous aldosterone infusion. Rocha et al. [45] reported similar findings in salt-loaded stroke-prone spontaneously hypertensive rats (SHR). Indeed, proteinuria is enhanced in aldosterone excess states, such as primary aldosteronism and rats infused with aldosterone [29, 30, 46, 47]. Conversely, aldosterone blockers effectively ameliorate proteinuria in patients or animal models of hypertension, diabetes, and renal disease [10, 12, 48–53]. Moreover, enhanced aldosterone signaling is found in the kidney biopsies from patients with heavy proteinuria [54]. The pivotal role for Sgk1 in the pathogenesis of proteinuria is also suggested by the report that gene targeting of Sgk1 protects against deoxycorticosterone-acetate/salt-induced albuminuria [55, 56]. However, the molecular mechanisms by which aldosterone enhances proteinuria had not been clearly elucidated until recently.
Glomerular podocyte injury as a major cause of proteinuria
The glomerular filtration barrier to plasma macromolecules comprises three layers: the fenestrated capillary endothelium, the glomerular basement membrane, and the podocytes. Podocytes line the outer aspect of the glomerular basement membrane, and serve as the final defense against urinary protein loss in the process of primary urine formation [57–59]. In 1998, Kestila et al. [60] identified nephrin as a causative gene for congenital nephrotic syndrome of the Finnish type using positional cloning. Nephrin is localized to the slit diaphragm, a unique apparatus formed at the junction of the interdigitating foot processes of podocytes, and thought to constitute the major size-selective permeability barrier. Since then, a number of podocyte-specific and slit diaphragm-specific molecules have been identified, including CD2AP, podocin, and α-actinin 4 [61–63]. Genetic mutations or gene targeting of many of these molecules are reported to manifest proteinuria and glomerulosclerosis, suggesting the pivotal roles of podocytes and their slit diaphragm as a filtration barrier [57]. Podocytes are actually injured in many types of proteinuric renal diseases, including nephrotic syndrome, lupus nephritis, diabetic and hypertensive nephropathy, and obesity-related glomerulopathy [12, 64–67]. It is also noteworthy that podocytes are terminally-differentiated cells and do not typically proliferate in response to injury. Once damaged, podocytopenia follows, ultimately culminating in glomerulosclerosis. It is podocyte damage, not mesangial abnormality, that triggers subsequent glomerular sclerosis [66]. Thus, podocytes can be an important therapeutic target.
Podocytes express a variety of vasoactive substances and their receptors, including components of the renin-angiotensin system, catecholamines, endothelins, natriuretic peptides, adrenomedullin, and nitric oxide [68–73]. From the anatomical aspect, podocytes, whose foot processes overlay on the glomerular capillary tufts, are quite sensitive to mechanical stress. For example, cyclic mechanical strain was shown to increase Ang II and its receptors in the podocytes [74]. Thus, podocyte functions may be modulated by the direct actions of vasoactive factors. However, the role of aldosterone had not been examined.
Aldosterone evokes podocyte injury: role of oxidative stress
The intimate relationship between aldosterone and proteinuria prompted us to investigate the effects of aldosterone on glomerular podocytes. We first analyzed proteinuria and podocyte injury in uninephrectomized rats continuously infused with aldosterone (0.75 μg/h via an osmotic minipump) and fed a high salt diet [11]. After 4 weeks, aldosterone-infused rats developed hypertension (systolic BP 214 ± 5 mm Hg vs 130 ± 3 mm Hg in control group, P < 0.01) and massive proteinuria (335 ± 42 mg/day vs 16 ± 2 mg/day in control group, P < 0.01). Glomerular expressions of slit diaphragm-associated molecules nephrin and podocin were markedly decreased, whereas expression of a damaged podocyte marker desmin [75] was upregulated in aldosterone-infused rats. Electron microscopic analysis revealed podocyte foot process effacement. Podocytes were already impaired at 2 weeks of aldosterone infusion, when proteinuria was modestly increased (37 ± 12 mg/day). Proteinuria and podocyte damage were completely reversed by selective aldosterone blocker eplerenone. These findings suggest that podocyte injury underlies the pathogenesis of proteinuria caused by aldosterone administration.
Then how does aldosterone evoke podocyte injury? In order to test the possible contribution of BP elevation per se, some aldosterone-infused rats were administered with hydralazine. Although hydralazine treatment normalized BP, it failed to improve proteinuria and podocyte damage, suggesting the presence of BP-independent mechanisms. We next examined the role of oxidative stress, because ROS are proposed as important mediators of aldosterone-induced target organ injury, as mentioned above. Aldosterone-infused rats exhibited enhanced oxidative stress markers, such as increased urinary excretion of thiobarbituric acid reactive substances, elevated renal NADPH oxidase activity determined using lucigenin chemiluminescence assay, and stimulation of membrane translocation of NADPH oxidase cytosolic components p67phox and Rac1. Indeed, an antioxidant tempol significantly reduced oxidative stress markers and corrected podocyte damage and proteinuria (53 ± 16 mg/day in tempol-treated group vs 245 ± 23 mg/day in untreated group, P < 0.01). Oxidative stress markers were also suppressed by eplerenone. These results indicate that aldosterone causes podocyte injury and proteinuria, possibly through induction of oxidant stress.
Aldosterone may modulate podocyte function directly by acting on MR within podocytes, or indirectly by affecting glomerular hemodynamics. Therefore, we examined whether aldosterone exerts direct cellular effects using culture podocytes established by Mundel et al. [76]. MR transcripts and proteins were actually detected in vitro cultured podocytes as well as in vivo glomerular podocytes, although at lower levels than in the distal nephron. Exposure of cultured podocytes to aldosterone resulted in nuclear translocation of MR, activation of NADPH oxidase, and ROS increment. In addition, aldosterone upregulated the expression of an aldosterone effector kinase Sgk1 in cultured podocytes as well as in the kidney of aldosterone-infused rats, which was prevented by aldosterone blockers. Taken together, our results suggest that at least some of the proteinuric effects of aldosterone can be attributed to direct actions on podocytes, including induction of oxidative stress and Sgk1 upregulation.
Metabolic syndrome, proteinuria, and aldosterone
The recent epidemic of obesity, especially among adolescents, is a major crisis worldwide [77, 78]. Clinical studies revealed that obesity is an important independent risk factor for proteinuria, CKD, and end-stage renal disease [79–82]. Furthermore, massive obesity is reported to be complicated with obesity-related glomerulopathy with nephrotic-range proteinuria, glomerulomegaly, and focal and segmental glomerulosclerosis [83–86].
Metabolic syndrome, a constellation of comorbidities that include visceral obesity, hypertension, glucose intolerance, and dyslipidemia, is also increasing at alarming rate, and becomes a serious social issue [87, 88]. Metabolic syndrome is also shown to increase the risk for proteinuria and CKD, independent of diabetes or hypertension [89–92].
Aldosterone excess has been implicated in obesity-related disorders. Tuck et al. [93] first reported the involvement of aldosterone in the pathogenesis of obesity hypertension. Several groups indicated that weight reduction in obese hypertensives decreased plasma aldosterone and BP [94, 95]. de Paula et al. [96] reported that diet-induced obesity in dogs was accompanied by elevated aldosterone, and that eplerenone effectively reduced diet-induced BP increment. Bochud et al. [97] recently reported that plasma aldosterone is independently associated with the metabolic syndrome. Based on these findings, we postulated a hypothesis that endogenous aldosterone contributes to podocyte injury, proteinuria, and CKD in metabolic syndrome.
Role of aldosterone in the early nephropathy of rats with metabolic syndrome
We used SHR/NDmcr-cp (SHR/cp) as a rat model of metabolic syndrome [13]. This rat is a derivative of SHR with spontaneous mutation in the leptin receptor gene, [98] and manifested a clustering of obesity, hypertension, hyperinsulinemia, hypertriglyceridemia, and elevated free fatty acids, fulfilling the criteria of metabolic syndrome (Fig. 1a) [87, 88]. Urinary protein excretion remained low in non-obese SHR. By contrast, the metabolic syndrome model SHR/cp showed exaggerated proteinuria despite similar BP elevation (Fig. 1b). Proteinuria in SHR/cp was accompanied by podocyte injury, as indicated by attenuation of normal podocyte marker nephrin, induction of injured podocyte marker desmin, and foot process effacement (Fig. 1c). These findings suggest that podocyte injury underlies the etiology of proteinuria in SHR/cp.

Metabolic syndrome model SHR/cp exhibited enhanced proteinuria and glomerular podocyte injury, which were improved by eplerenone. a External appearance of 17-week-old SHR and SHR/cp. b Time course of proteinuria in SHR and SHR/cp. c Transmission electron microscopy of glomeruli from 17-week-old SHR and SHR/cp. d Effect of eplerenone on proteinuria in SHR/cp
Notably, serum aldosterone level was higher in SHR/cp than in non-obese SHR, and there was a positive correlation between circulating aldosterone concentration and proteinuria. Expression of Sgk1, one of the downstream targets of aldosterone signaling, was also significantly upregulated in the whole kidney as well as the glomerular fraction of SHR/cp. We thus administered eplerenone to SHR/cp. Eplerenone effectively reduced proteinuria in SHR/cp (Fig. 1d). In parallel, eplerenone improved podocyte injury in SHR/cp, as revealed by the changes in nephrin and desmin expressions. These data suggest that aldosterone-provoked podocyte injury plays an important role in the pathogenesis of proteinuria in SHR/cp.
We further explored the role of oxidative stress. Similar to the case of aldosterone-infused rats, oxidative stress markers were upregulated in SHR/cp. Importantly, eplerenone reversed the increased oxidative stress, suggesting the participation of endogenous aldosterone. Treatment with antioxidant tempol significantly alleviated proteinuria and podocyte injury in SHR/cp. These data suggest that oxidative stress is an important mediator of endogenous aldosterone-induced podocyte injury in SHR/cp.
Mechanisms of elevated serum aldosterone in SHR/cp: possible contribution of adipocyte-derived aldosterone releasing factors
We further investigated the mechanisms of high aldosterone state in SHR/cp. Expression of aldosterone synthase (CYP11B2) was enhanced in the adrenal glands of SHR/cp but was below the detection level in the kidney, suggesting that aldosterone production in the adrenals is responsible for high circulating aldosterone in SHR/cp. Aldosterone is synthesized and released from the zona glomerulosa cells of the adrenal cortex [99]. Plasma renin-angiotensin system is a major regulator of adrenal aldosterone production. However, SHR/cp displayed lower plasma renin activity, suggesting that this is not the responsible factor. Hyperkalemia is another well-known aldosterone secretagogue, but this was not the case in SHR/cp, either. Several reports showed that plasma aldosterone in obese or metabolic syndrome patients was not associated with plasma renin but with the amount of visceral fat [97, 100]. Adipose tissue is now recognized as a dynamic endocrine organ secreting a variety of adipokines [101, 102]. To our interest, Ehrhart-Bornstein et al. [103] reported adipocyte-derived substances that stimulate adrenal aldosterone production. Scanning electron micrographs showed markedly enlarged adipocytes in SHR/cp, implicating the pathogenetic role of adipocyte-derived factors.
So, we compared the aldosterone-releasing activity of adipocytes between SHR/cp and SHR. We cultured isolated adipocytes and collected fat cell conditioned medium (FCCM), which contains adipocyte-derived factors. Indeed, aldosterone production in H295R adrenocortical cells was markedly increased by FCCM from SHR/cp but not that from non-obese SHR. The activity was not recapitulated by known adipocytokines. The aldosterone releasing activity of FCCM was not inhibited by ARB, excluding the contribution of Ang II. In addition, angiotensinogen mRNA expression in the adipose tissues was not elevated in SHR/cp. In parallel, FCCM from SHR/cp but not from SHR stimulated the expression of aldosterone synthase and steroidogenic acute regulatory protein, another key factor in aldosterone synthesis that mediates transfer of cholesterol to mitochondria, [104] in H295R cells. These findings suggest the involvement of adipocyte-derived substances other than Ang II in hyperaldosteronism in SHR/cp.
Goodfriend et al. [105] reported that epoxy-keto derivative of linoleic acid, one of the oxidized products of fatty acids, stimulates aldosterone secretion in rat adrenal cells. Although they originally hypothesized that the site of oxidative modification might be the liver, adipocytes might also contribute to the epoxy-keto modification of linoleic acid.
It should be noted that hyperaldosteronism caused by these adipocyte-derived factors is not inhibitable by ACEIs or ARBs. Thus, eplerenone should have benefit over Ang II blockade in situations where such factors are overproduced. We expect that these adipocyte-derived aldosterone releasing factors, if identified, can be a novel target of therapy in the metabolic syndrome. These substances might have a pathogenetic role also in end-stage renal disease, a condition with renin-independent hyperaldosteronism [106].
Taken together, our data indicate that adipocytes produce aldosterone releasing factors in SHR/cp, which might contribute to elevated circulating aldosterone, podocyte injury and proteinuria in this model.
Salt accelerates nephropathy in SHR/cp: involvement of MR activation
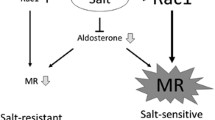
Next, we investigated the effect of salt loading on renal damage in SHR/cp, because high sodium intake is known to exacerbate proteinuria in obese subjects [107]. SHR/cp fed a high sodium diet for 4 weeks developed massive proteinuria and advanced renal lesions including glomerular podocyte impairment (proteinuria: 260 ± 64 mg/day in salt-loaded SHR/cp vs 44 ± 20 mg/day in non-salt-loaded group, P < 0.01). Surprisingly, eplerenone dramatically ameliorated the salt-induced proteinuria and renal injury in SHR/cp (proteinuria: 36 ± 3 mg/day in salt-loaded SHR/cp treated with eplerenone; Fig. 2a) [14]. Salt-induced nephropathy in SHR/cp was accompanied by renal overexpression of aldosterone-dependent genes, such as PAI-1, MCP-1, and TGF-β1, which were all corrected by eplerenone. Although salt loading reduced circulating aldosterone possibly due to volume overload, it paradoxically increased nuclear MR and expression of Sgk1 in the kidney (Fig. 2b, c). To clarify the discrepancy between decreased aldosterone and enhanced renal MR signaling by salt, we further investigated the role of oxidative stress, a putative key factor mediating salt-induced tissue damage [108, 109]. Interestingly, antioxidant tempol attenuated the salt-evoked MR upregulation and Sgk1 induction, and alleviated proteinuria and renal histologic abnormalities (Fig. 2d, e). Nuclear MR and expression of Sgk1 were pronounced in the kidneys of SHR/cp compared with those of SHR. Taken together, MR activation in the kidney plays a pivotal role in the pathogenesis of salt-induced nephropathy in SHR/cp, and oxidative stress, at least partially, contributes to the salt-evoked MR activation [14].

Salt-induced nephropathy in SHR/cp and roles of renal MR activation and oxidative stress. a High salt diet markedly increased proteinuria in SHR/cp, which was dramatically normalized by eplerenone. b High salt lowered serum aldosterone concentration. c High salt caused MR activation in the kidney, as evidenced by nuclear MR content and renal Sgk1 expression. d Salt-induced MR activation in SHR/cp was inhibited by antioxidant Tempol. e Tempol also improved proteinuria in salt-loaded SHR/cp
The comparison between SHR and SHR/cp suggests that some factor(s) related to obesity might be involved in the increased salt-sensitivity of renal injury in SHR/cp. Although high salt diet reduced circulating aldosterone in SHR/cp, this inhibition was blunted in this model as compared with salt-resistant animals. The above-mentioned adipocyte-derived aldosterone releasing factors is angiotensin II-independent, so should not be suppressed by salt loading. As a result, suppression of circulating aldosterone was less than expected for the amount of salt intake, which might be another mechanism of salt-sensitive renal injury in this model.
Aldosterone and podocyte injury in salt-hypertensive model
We further examined the effect of eplerenone on podocytes in Dahl salt-hypertensive rats that have relatively low circulating aldosterone levels. Podocyte injury was detected as early as 2 weeks after salt loading, when proteinuria was only modestly increased. Treatment with eplerenone or hydralazine reduced BP to the similar extent. However, only eplerenone normalized podocyte damage and retarded the progression of proteinuria and glomerulosclerosis in this model. Renal MR signaling was actually reported to be enhanced in the kidneys of Dahl salt hypertensive rats [110].
Conclusions and perspectives
We reviewed the novel role of aldosterone as a mediator of glomerular podocyte injury. Oxidative stress was suggested to be involved in this process. Aldosterone blockers are expected to confer renoprotection in high aldosterone states such as primary aldosteronism, some patients with metabolic syndrome, renal insufficiency, and heavy proteinuria. Our results implicate that aldosterone blockers can also be renoprotective in patients with activated MR signaling in the target tissue, whose circulating aldosterone level is not necessarily high, as is seen in metabolic syndrome rats fed a high salt diet. In fact, aldosterone blockers effectively reduce BP in low renin hypertensive patients and patients with high salt intake. Addition of aldosterone blockers in patients treated with ACEIs or ARBs are also successful, because 45–50% of patients treated with ACEIs and 25% of those with ARBs have elevated aldosterone concentrations because of “aldosterone breakthrough”. In Japan, eplerenone became available for general practitioners in autumn 2007. It should be noted that we have to be careful about hyperkalemia, especially in patients with impaired renal function. To our regret, administration of eplerenone is contraindication for diabetic patients with albuminuria at present, because some studies showed high occurrence of hyperkalemia, although animal studies indicated potent renoprotective effects of aldosterone blockers in diabetic nephropathy. We believe that aldosterone blockers with anti-proteinuric and podocyte-protective properties are promising drugs for the prevention and treatment of target organ injury in life style-related illnesses.
References
Simpson SA, Tait JF, Wettstein A, Neher R, von Euw J, Reichstein T. Isolierung eines neuen kristallisierten Hormons aus Nebennerien mit besonders hoher Wirksamkeit auf den Mineralsoffwechsel. Experientia. 1953;9:333–5.
Williams JS, Williams GH. 50th anniversary of aldosterone. J Clin Endocrinol Metab. 2003;88:2364–72.
Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–56.
Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, et al. Human hypertension caused by mutations in WNK kinases. Science. 2001;293:1107–12.
Hostetter TH, Ibrahim HN. Aldosterone in chronic kidney and cardiac disease. J Am Soc Nephrol. 2003;14:2395–401.
Rocha R, Funder JW. The pathophysiology of aldosterone in the cardiovascular system. Ann NY Acad Sci. 2002;970:89–100.
Fuller PJ, Young MJ. Mechanisms of mineralocorticoid action. Hypertension. 2005;46:1227–35.
Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized aldactone evaluation study investigators. N Engl J Med. 1999;341:709–17.
Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309–21.
Epstein M. Aldosterone blockade: an emerging strategy for abrogating progressive renal disease. Am J Med. 2006;119:912–9.
Shibata S, Nagase M, Yoshida S, Kawachi H, Fujita T. Podocyte as the target for aldosterone: roles of oxidative stress and Sgk1. Hypertension. 2007;49:355–64.
Nagase M, Shibata S, Yoshida S, Nagase T, Gotoda T, Fujita T. Podocyte injury underlies the glomerulopathy of Dahl salt-hypertensive rats and is reversed by aldosterone blocker. Hypertension. 2006;47:1084–93.
Nagase M, Yoshida S, Shibata S, Nagase T, Gotoda T, Ando K, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol. 2006;17:3438–46.
Nagase M, Matsui H, Shibata S, Gotoda T, Fujita T. Salt-induced nephropathy in obese spontaneously hypertensive rats via paradoxical activation of the mineralocorticoid receptor: role of oxidative stress. Hypertension. 2007;50:877–83.
Rozansky DJ. The role of aldosterone in renal sodium transport. Semin Nephrol. 2006;26:173–81.
Meneton P, Loffing J, Warnock DG. Sodium and potassium handling by the aldosterone-sensitive distal nephron: the pivotal role of the distal and connecting tubule. Am J Physiol Renal Physiol. 2004;287:F593–601.
Knepper MA, Kim GH, Masilamani S. Renal tubule sodium transporter abundance profiling in rat kidney: response to aldosterone and variations in NaCl intake. Ann NY Acad Sci. 2003;986:562–9.
Lang F, Bohmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V. (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev. 2006;86:1151–78.
Hou J, Speirs HJ, Seckl JR, Brown RW. Sgk1 gene expression in kidney and its regulation by aldosterone: spatio-temporal heterogeneity and quantitative analysis. J Am Soc Nephrol. 2002;13:1190–8.
Ring AM, Leng Q, Rinehart J, Wilson FH, Kahle KT, Hebert SC, et al. An SGK1 site in WNK4 regulates Na+ channel and K+ channel activity and has implications for aldosterone signaling and K+ homeostasis. Proc Natl Acad Sci USA. 2007;104:4025–9.
Berger S, Bleich M, Schmid W, Cole TJ, Peters J, Watanabe H, et al. Mineralocorticoid receptor knockout mice: pathophysiology of Na+ metabolism. Proc Natl Acad Sci USA. 1998;95:9424–9.
Ronzaud C, Loffing J, Bleich M, Gretz N, Grone HJ, Schutz G, et al. Impairment of sodium balance in mice deficient in renal principal cell mineralocorticoid receptor. J Am Soc Nephrol. 2007;18:1679–87.
Hummler E, Barker P, Talbot C, Wang Q, Verdumo C, Grubb B, et al. A mouse model for the renal salt-wasting syndrome pseudohypoaldosteronism. Proc Natl Acad Sci USA. 1997;94:11710–5.
Pradervand S, Barker PM, Wang Q, Ernst SA, Beermann F, Grubb BR, et al. Salt restriction induces pseudohypoaldosteronism type 1 in mice expressing low levels of the beta-subunit of the amiloride-sensitive epithelial sodium channel. Proc Natl Acad Sci USA. 1999;96:1732–7.
Wulff P, Vallon V, Huang DY, Volkl H, Yu F, Richter K, et al. Impaired renal Na(+) retention in the sgk1-knockout mouse. J Clin Invest. 2002;110:1263–8.
Epstein M. Aldosterone and the hypertensive kidney: its emerging role as a mediator of progressive renal dysfunction: a paradigm shift. J Hypertens. 2001;19:829–42.
Rocha R, Rudolph AE, Frierdich GE, Nachowiak DA, Kekec BK, Blomme EA, et al. Aldosterone induces a vascular inflammatory phenotype in the rat heart. Am J Physiol Heart Circ Physiol. 2002;283:H1802–10.
Wehling M. Specific, nongenomic actions of steroid hormones. Annu Rev Physiol. 1997;59:365–93.
Blasi ER, Rocha R, Rudolph AE, Blomme EA, Polly ML, McMahon EG. Aldosterone/salt induces renal inflammation and fibrosis in hypertensive rats. Kidney Int. 2003;63:1791–800.
Nishiyama A, Yao L, Nagai Y, Miyata K, Yoshizumi M, Kagami S, et al. Possible contributions of reactive oxygen species and mitogen-activated protein kinase to renal injury in aldosterone/salt-induced hypertensive rats. Hypertension. 2004;43:841–8.
Miyata K, Rahman M, Shokoji T, Nagai Y, Zhang GX, Sun GP, et al. Aldosterone stimulates reactive oxygen species production through activation of NADPH oxidase in rat mesangial cells. J Am Soc Nephrol. 2005;16:2906–12.
Terada Y, Kobayashi T, Kuwana H, Tanaka H, Inoshita S, Kuwahara M, et al. Aldosterone stimulates proliferation of mesangial cells by activating mitogen-activated protein kinase 1/2, cyclin D1, and cyclin A. J Am Soc Nephrol. 2005;16:2296–305.
Nagai Y, Miyata K, Sun GP, Rahman M, Kimura S, Miyatake A, et al. Aldosterone stimulates collagen gene expression and synthesis via activation of ERK1/2 in rat renal fibroblasts. Hypertension. 2005;46:1039–45.
Ishizawa K, Izawa Y, Ito H, Miki C, Miyata K, Fujita Y, et al. Aldosterone stimulates vascular smooth muscle cell proliferation via big mitogen-activated protein kinase 1 activation. Hypertension. 2005;46:1046–52.
Sun Y, Zhang J, Lu L, Chen SS, Quinn MT, Weber KT. Aldosterone-induced inflammation in the rat heart : role of oxidative stress. Am J Pathol. 2002;161:1773–81.
Keidar S, Kaplan M, Pavlotzky E, Coleman R, Hayek T, Hamoud S, et al. Aldosterone administration to mice stimulates macrophage NADPH oxidase and increases atherosclerosis development: a possible role for angiotensin-converting enzyme and the receptors for angiotensin II and aldosterone. Circulation. 2004;109:2213–20.
Leopold JA, Dam A, Maron BA, Scribner AW, Liao R, Handy DE, et al. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med. 2007;13:189–97.
Nishiyama A, Yao L, Fan Y, Kyaw M, Kataoka N, Hashimoto K, et al. Involvement of aldosterone and mineralocorticoid receptors in rat mesangial cell proliferation and deformability. Hypertension. 2005;45:710–6.
Sun GP, Kohno M, Guo P, Nagai Y, Miyata K, Fan YY, et al. Involvements of Rho-kinase and TGF-beta pathways in aldosterone-induced renal injury. J Am Soc Nephrol. 2006;17:2193–201.
K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 2002;39:S1–266.
Ibrahim HN, Hostetter TH. Aldosterone in renal disease. Curr Opin Nephrol Hypertens. 2003;12:159–64.
Brown NJ. Aldosterone and end-organ damage. Curr Opin Nephrol Hypertens. 2005;14:235–41.
Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The collaborative study group. N Engl J Med. 1993;329:1456–62.
Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest. 1996;98:1063–8.
Rocha R, Chander PN, Zuckerman A, Stier CT Jr. Role of aldosterone in renal vascular injury in stroke-prone hypertensive rats. Hypertension. 1999;33:232–7.
Ribstein J, Du Cailar G, Fesler P, Mimran A. Relative glomerular hyperfiltration in primary aldosteronism. J Am Soc Nephrol. 2005;16:1320–5.
Rossi GP, Bernini G, Desideri G, Fabris B, Ferri C, Giacchetti G, et al. Renal damage in primary aldosteronism: results of the PAPY Study. Hypertension. 2006;48:232–8.
White WB, Duprez D, St Hillaire R, Krause S, Roniker B, Kuse-Hamilton J, et al. Effects of the selective aldosterone blocker eplerenone versus the calcium antagonist amlodipine in systolic hypertension. Hypertension. 2003;41:1021–6.
Williams GH, Burgess E, Kolloch RE, Ruilope LM, Niegowska J, Kipnes MS, et al. Efficacy of eplerenone versus enalapril as monotherapy in systemic hypertension. Am J Cardiol. 2004;93:990–6.
Sato A, Hayashi K, Naruse M, Saruta T. Effectiveness of aldosterone blockade in patients with diabetic nephropathy. Hypertension. 2003;41:64–8.
Han SY, Kim CH, Kim HS, Jee YH, Song HK, Lee MH, et al. Spironolactone prevents diabetic nephropathy through an anti-inflammatory mechanism in type 2 diabetic rats. J Am Soc Nephrol. 2006;17:1362–72.
Schjoedt KJ, Rossing K, Juhl TR, Boomsma F, Tarnow L, Rossing P, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int. 2006;70:536–42.
Chrysostomou A, Becker G. Spironolactone in addition to ACE inhibition to reduce proteinuria in patients with chronic renal disease. N Engl J Med. 2001;345:925–6.
Quinkler M, Zehnder D, Eardley KS, Lepenies J, Howie AJ, Hughes SV, et al. Increased expression of mineralocorticoid effector mechanisms in kidney biopsies of patients with heavy proteinuria. Circulation. 2005;112:1435–43.
Artunc F, Amann K, Nasir O, Friedrich B, Sandulache D, Jahovic N, et al. Blunted DOCA/high salt induced albuminuria and renal tubulointerstitial damage in gene-targeted mice lacking SGK1. J Mol Med. 2006;84:737–46.
Vallon V, Huang DY, Grahammer F, Wyatt AW, Osswald H, Wulff P, et al. SGK1 as a determinant of kidney function and salt intake in response to mineralocorticoid excess. Am J Physiol Regul Integr Comp Physiol. 2005;289:R395-401.
Tryggvason K, Patrakka J, Wartiovaara J. Hereditary proteinuria syndromes and mechanisms of proteinuria. N Engl J Med. 2006;354:1387–401.
Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253–307.
Reiser J, Mundel P. Danger signaling by glomerular podocytes defines a novel function of inducible B7–1 in the pathogenesis of nephrotic syndrome. J Am Soc Nephrol. 2004;15:2246–8.
Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, et al. Positionally cloned gene for a novel glomerular protein–nephrin–is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575–82.
Shih NY, Li J, Karpitskii V, Nguyen A, Dustin ML, Kanagawa O, et al. Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science. 1999;286:312–5.
Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet. 2000;24:349–54.
Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet. 2000;24:251–6.
Pagtalunan ME, Miller PL, Jumping-Eagle S, Nelson RG, Myers BD, Rennke HG, et al. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest. 1997;99:342–8.
Wolf G, Chen S, Ziyadeh FN. From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes. 2005;54:1626–34.
Kretzler M, Koeppen-Hagemann I, Kriz W. Podocyte damage is a critical step in the development of glomerulosclerosis in the uninephrectomised-desoxycorticosterone hypertensive rat. Virchows Arch. 1994;425:181–93.
Chen HM, Liu ZH, Zeng CH, Li SJ, Wang QW, Li LS. Podocyte lesions in patients with obesity-related glomerulopathy. Am J Kidney Dis. 2006;48:772–9.
Sharma M, Sharma R, Greene AS, McCarthy ET, Savin VJ. Documentation of angiotensin II receptors in glomerular epithelial cells. Am J Physiol. 1998;274:F623–7.
Huber TB, Gloy J, Henger A, Schollmeyer P, Greger R, Mundel P, et al. Catecholamines modulate podocyte function. J Am Soc Nephrol. 1998;9:335–45.
Ortmann J, Amann K, Brandes RP, Kretzler M, Munter K, Parekh N, et al. Role of podocytes for reversal of glomerulosclerosis and proteinuria in the aging kidney after endothelin inhibition. Hypertension. 2004;44:974–81.
Lewko B, Endlich N, Kriz W, Stepinski J, Endlich K. C-type natriuretic peptide as a podocyte hormone and modulation of its cGMP production by glucose and mechanical stress. Kidney Int. 2004;66:1001–8.
Nagase M, Ando K, Katafuchi T, Kato A, Hirose S, Fujita T. Role of natriuretic peptide receptor type C in Dahl salt-sensitive hypertensive rats. Hypertension. 1997;30:177–83.
Hino M, Nagase M, Kaname S, Shibata S, Nagase T, Oba S, et al. Expression and regulation of adrenomedullin in renal glomerular podocytes. Biochem Biophys Res Commun. 2005;330:178–85.
Durvasula RV, Petermann AT, Hiromura K, Blonski M, Pippin J, Mundel P, et al. Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int. 2004;65:30–9.
Yaoita E, Kawasaki K, Yamamoto T, Kihara I. Variable expression of desmin in rat glomerular epithelial cells. Am J Pathol. 1990;136:899–908.
Mundel P, Reiser J, Zuniga Mejia Borja A, Pavenstadt H, Davidson GR, Kriz W, et al. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res. 1997;236:248–58.
Mann CC. Public health. Provocative study says obesity may reduce U S life expectancy. Science. 2005;307:1716–7.
Flier JS. Obesity wars: molecular progress confronts an expanding epidemic. Cell. 2004;116:337–50.
Fox CS, Larson MG, Leip EP, Culleton B, Wilson PW, Levy D. Predictors of new-onset kidney disease in a community-based population. Jama. 2004;291:844–50.
Hsu CY, McCulloch CE, Iribarren C, Darbinian J, Go AS. Body mass index and risk for end-stage renal disease. Ann Intern Med. 2006;144:21–8.
Iseki K, Ikemiya Y, Kinjo K, Inoue T, Iseki C, Takishita S. Body mass index and the risk of development of end-stage renal disease in a screened cohort. Kidney Int. 2004;65:1870–6.
Gelber RP, Kurth T, Kausz AT, Manson JE, Buring JE, Levey AS, et al. Association between body mass index and CKD in apparently healthy men. Am J Kidney Dis. 2005;46:871–80.
Weisinger JR, Kempson RL, Eldridge FL, Swenson RS. The nephrotic syndrome: a complication of massive obesity. Ann Intern Med. 1974;81:440–7.
Adelman RD. Obesity and renal disease. Curr Opin Nephrol Hypertens. 2002;11:331–5.
Henegar JR, Bigler SA, Henegar LK, Tyagi SC, Hall JE. Functional and structural changes in the kidney in the early stages of obesity. J Am Soc Nephrol. 2001;12:1211–7.
Praga M, Morales E. Obesity, proteinuria and progression of renal failure. Curr Opin Nephrol Hypertens. 2006;15:481–6.
Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112:2735–52.
Alberti KG, Zimmet P, Shaw J. The metabolic syndrome--a new worldwide definition. Lancet. 2005;366:1059–62.
Chen J, Muntner P, Hamm LL, Jones DW, Batuman V, Fonseca V, et al. The metabolic syndrome and chronic kidney disease in U.S. adults. Ann Intern Med. 2004;140:167–74.
Kurella M, Lo JC, Chertow GM. Metabolic syndrome and the risk for chronic kidney disease among nondiabetic adults. J Am Soc Nephrol. 2005;16:2134–40.
Tanaka H, Shiohira Y, Uezu Y, Higa A, Iseki K. Metabolic syndrome and chronic kidney disease in Okinawa, Japan. Kidney Int. 2006;69:369–74.
Ninomiya T, Kiyohara Y, Kubo M, Yonemoto K, Tanizaki Y, Doi Y, et al. Metabolic syndrome and CKD in a general Japanese population: the Hisayama Study. Am J Kidney Dis. 2006;48:383–91.
Tuck ML, Sowers J, Dornfeld L, Kledzik G, Maxwell M. The effect of weight reduction on blood pressure, plasma renin activity, and plasma aldosterone levels in obese patients. N Engl J Med. 1981;304:930–3.
Engeli S, Bohnke J, Gorzelniak K, Janke J, Schling P, Bader M, et al. Weight loss and the renin-angiotensin-aldosterone system. Hypertension. 2005;45:356–62.
Ruano M, Silvestre V, Castro R, Garcia-Lescun MC, Rodriguez A, Marco A, et al. Morbid obesity, hypertensive disease and the renin-angiotensin-aldosterone axis. Obes Surg. 2005;15:670–6.
de Paula RB, da Silva AA, Hall JE. Aldosterone antagonism attenuates obesity-induced hypertension and glomerular hyperfiltration. Hypertension. 2004;43:41–7.
Bochud M, Nussberger J, Bovet P, Maillard MR, Elston RC, Paccaud F, et al. Plasma aldosterone is independently associated with the metabolic syndrome. Hypertension. 2006;48:239–45.
Takaya K, Ogawa Y, Hiraoka J, Hosoda K, Yamori Y, Nakao K, et al. Nonsense mutation of leptin receptor in the obese spontaneously hypertensive Koletsky rat. Nat Genet. 1996;14:130–1.
Williams GH. Aldosterone biosynthesis, regulation, and classical mechanism of action. Heart Fail Rev. 2005;10:7–13.
Goodfriend TL, Calhoun DA. Resistant hypertension, obesity, sleep apnea, and aldosterone: theory and therapy. Hypertension. 2004;43:518–24.
Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91.
Shimomura I, Funahashi T, Takahashi M, Maeda K, Kotani K, Nakamura T, et al. Enhanced expression of PAI-1 in visceral fat: possible contributor to vascular disease in obesity. Nat Med. 1996;2:800–3.
Ehrhart-Bornstein M, Lamounier-Zepter V, Schraven A, Langenbach J, Willenberg HS, Barthel A, et al. Human adipocytes secrete mineralocorticoid-releasing factors. Proc Natl Acad Sci USA. 2003;100:14211–6.
Stocco DM, Wang X, Jo Y, Manna PR. Multiple signaling pathways regulating steroidogenesis and steroidogenic acute regulatory protein expression: more complicated than we thought. Mol Endocrinol. 2005;19:2647–59.
Goodfriend TL, Ball DL, Egan BM, Campbell WB, Nithipatikom K. Epoxy-keto derivative of linoleic acid stimulates aldosterone secretion. Hypertension. 2004;43:358–63.
Hene RJ, Boer P, Koomans HA, Mees EJ. Plasma aldosterone concentrations in chronic renal disease. Kidney Int. 1982;21:98–101.
Verhave JC, Hillege HL, Burgerhof JG, Janssen WM, Gansevoort RT, Navis GJ, et al. Sodium intake affects urinary albumin excretion especially in overweight subjects. J Intern Med. 2004;256:324–30.
Ritz E, Dikow R, Morath C, Schwenger V. Salt—a potential “uremic toxin?” Blood Purif. 2006;24:63–6.
Kitiyakara C, Chabrashvili T, Chen Y, Blau J, Karber A, Aslam S, et al. Salt intake, oxidative stress, and renal expression of NADPH oxidase and superoxide dismutase. J Am Soc Nephrol. 2003;14:2775–82.
Farjah M, Roxas BP, Geenen DL, Danziger RS. Dietary salt regulates renal SGK1 abundance: relevance to salt sensitivity in the Dahl rat. Hypertension. 2003;41:874–8.
Author information
Authors and Affiliations
Corresponding author
Additional information
Presented at the 50th annual meeting of the Japanese Society of Nephrology.
About this article
Cite this article
Nagase, M., Fujita, T. Aldosterone and glomerular podocyte injury. Clin Exp Nephrol 12, 233–242 (2008). https://doi.org/10.1007/s10157-008-0034-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10157-008-0034-9