
Abstract
Background
TAS-102 improved the overall survival of metastatic colorectal cancer (CRC) patients with a median progression-free survival (PFS) in the RECOURSE trial. Subsequently, the combination of TAS-102 and bevacizumab was shown to extend the median PFS (C-TASK FORCE study). However, the study included patients who received second- and third-line treatment. Our study exclusively examined patients receiving this combination as a third-line treatment to investigate the clinical impact beyond cytotoxic doublets.
Methods
This investigator-initiated, open-label, single-arm, multi-centered phase II study was conducted in Japan. Eligible CRC patients were refractory or intolerant to fluoropyrimidine, irinotecan, and oxaliplatin in first- and second-line therapy. TAS-102 (35 mg/m2) was given orally twice daily on days 1–5 and 8–12 in a 4-week cycle, and bevacizumab (5 mg/kg) was administered by intravenous infusion every 2 weeks. The primary endpoint was PFS and the secondary endpoints were time-to-treatment failure, response rate, overall survival (OS), and safety.
Results
Between June 2016 and August 2017, 32 patients were enrolled. All patients previously received bevacizumab. The median PFS was 4.5 months; the median overall survival was 9.3 months. Partial response was observed in two patients. The most common adverse events above grade 3 were neutropenia followed by thrombocytopenia. There were no non-hematological adverse events above grade 3 and no treatment-related deaths occurred.
Conclusions
This study met its primary endpoint of PFS, which is comparable to the results of the C-TASK FORCE study. The TAS-102 and bevacizumab combination has the potential to be a therapeutic option for third-line treatment of metastatic CRC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC), which killed approximately 880,000 people worldwide in 2018, is the leading cause of cancer death [1]. The response rate of metastatic CRC is approximately 50% for the first treatment but drops to 10–20% for the second treatment. In third-line treatment for CRC, chemotherapeutic drugs show little efficacy and tumor shrinkage is rarely observed [2]. Therefore, the goal of third-line treatment is to prolong survival and prevent tumor progression without compromising quality of life.
TAS-102 is an oral anticancer agent comprising trifluridine (FTD) and tipiracil hydrochloride [3]. Mechanistically, FTD incorporates into the DNA of CRC cells to exert its antitumor effect [4], whereas tipiracil hydrochloride maintains the blood concentration of FTD by inhibiting the FTD-degrading enzyme thymidine phosphorylase. In the global phase III RECOURSE trial, progression-free survival (PFS) and overall survival (OS) were significantly better in patients with CRC treated with TAS-102 than in those treated with placebo. The combination of TAS-102 and bevacizumab has been shown to extend the median PFS by 3.7 months (in the C-TASK FORCE study) [5] and 4.6 months (in a Danish trial) [6]. However, these studies included patients with second- and third-line treatment. There have been several reports of TAS-102 plus bevacizumab therapy in retrospective studies [7,8,9,10], but only two prospective studies, the C-TASK FORCE study and the Danish trial, have been published [5, 6].
Anti-VEGF antibody bevacizumab improves PFS and OS when added to first- and second-line treatment in metastatic CRC [11]. However, in patients who have already received bevacizumab as first- or second-line therapy, anti-EGFR antibodies are commonly given as third-line therapy, and the survival benefit of bevacizumab is unknown. The main purpose of this prospective study was to estimate the efficacy and safety of administration of TAS-102 plus bevacizumab as third-line treatment for patients with metastatic CRC.
Patients and methods
Patients
This study was designed as a prospective, investigator-initiated, non-randomized, single-arm, multi-centered open label phase II trial. Patients were recruited in 11 centers in Japan. Written informed consent was obtained from the patients prior to any screening or inclusion procedures. All procedures were performed in accordance with the Declaration of Helsinki. The trial was organized by the Department of Gastrointestinal and Hepato-Biliary-Pancreatic Surgery, Nippon Medical School, Tokyo, Japan.
This study was registered as UMIN000022438 on 1 August 2016. The estimated study completion date was January 2020 (final data collection and date of primary outcome measure).
Patient selection criteria
Inclusion criteria
-
1.
Pathologically proven CRC
-
2.
Unresectable primary or one or more unresectable metastatic tumor
-
3.
Previous administration of first- and second-line chemotherapy for metastatic CRC and tumors diagnosed as progression of disease (PD)
-
4.
Aged between 20 and 80 years
-
5.
Eastern Cooperative Oncology Group Performance Status (ECOG) of 0 or 1
-
6.
Measurable lesions based on the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1
-
7.
Able to take oral medication
-
8.
Life expectancy of at least 3 months
-
9.
Exhibiting sufficient organ function for up to 2 weeks prior to enrollment in the study with the following parameters considered:
-
•
Leukocyte count ≥ 3500/mm3
-
•
Absolute neutrophil count ≥ 1500/mm3
-
•
Hemoglobin level ≥ 8.0 g/dL
-
•
Platelet count ≥ 75,000/mm3
-
•
Total bilirubin level ≤ 1.5 mg/dL
-
•
Aspartate aminotransferase level ≤ 2.5 × upper limit of normal
-
•
Alanine aminotransferase level ≤ 2.5 × upper limit of normal
-
•
Serum creatinine level ≤ 1.5 mg/dL
-
•
No active infectious disease
-
•
No recognizable diarrhea or non-hematological adverse events (except for alopecia, dysgeusia, pigmentation)
-
10.
Signed written informed consent provided prior to enrollment in this study.
Exclusion criteria
-
1.
Contraindications for TAS-102 and bevacizumab
-
2.
History of treatment with TAS-102
-
3.
Severe drug allergy
-
4.
Severe liver dysfunction
-
5.
Women who were pregnant or planning a pregnancy and men intending to impregnate their partner
-
6.
Uncontrollable hypertension (systolic blood pressure ≥ 150 mmHg or diastolic blood pressure ≥ 90 mmHg, even when taking oral antihypertensives)
-
7.
Other serious complications (symptomatic unstable ischemic heart disease, arrhythmia, interstitial pneumonia, pulmonary fibrosis, renal failure, liver failure, uncontrollable diabetes mellitus, and gastrointestinal ulcers)
-
8.
Presence of other active cancers
-
9.
Clinical or radiological evidence of brain metastases
-
10.
Currently ongoing treatment with corticosteroids
-
11.
Any other criteria, for which the investigator deems patients unsuitable for this study
-
12.
Proteinuria ≥ grade 2
-
13.
Gastrointestinal ulcer or bleeding
-
14.
Previous hemoptysis
-
15.
Ongoing treatment with anticoagulant
-
16.
Synchronous or metachronous multiple malignancy within the last 5-year disease-free interval
Treatment
The TAS-CC3 study regimen comprised 28-day cycles of administration of TAS-102 (orally administered at a dose of 35 mg/m2 twice daily on days 1–5 and 8–12 of every 28-day cycle) plus bevacizumab (5.0 mg/kg on days 1 and 15).
Treatment was continued until any of the following occurred: progressive disease, consent withdrawal, unacceptable toxicity, discontinuation based on clinical indications, or at the investigator’s discretion. If dose reduction was required during treatment because of toxicity, the dose of TAS-102 was reduced in increments of 5 mg/m2. For neutropenia, TAS-102 was withdrawn at a neutrophil count of 1000/mm3 or less and the dose was reduced by 10 mg per day at a count of 500/mm3 or less. Dose reduction of bevacizumab was not recommended in principle. However, if patients had unacceptable toxicities related to bevacizumab, TAS-102 monotherapy could be continued according to the protocol without bevacizumab. Other chemotherapy, radiotherapy, immunotherapy, hormone therapy, and hyperthermia were prohibited during the trial. Prophylactic administration of G-CSF was prohibited, but there was no provision for its use in treatment for neutropenia. Clinical evaluations and CT scans were undertaken 8 (± 2) weeks after starting chemotherapy and then every 8 (± 2) weeks until progression. Response was determined by the local investigator at least 8 weeks after the first cycle according to the RECIST version 1.1.
Criteria for suspending and resuming drug administration
Adverse events were assessed using the Common Terminology Criteria for Adverse Events ver.4.0 (CTCAE ver.4.0). When neutropenia, thrombocytopenia, or hypertension was recognized, drug administration was suspended and resumed based on appropriate criteria. If other adverse events reached grade ≥ 3, drug administration was suspended until improvement to a grade ≤ 2 and then resumed with a 10 mg/day reduction in the TAS-102 dose. Additionally, attending physicians suspended or discontinued drug administration when deemed appropriate.
Study endpoints
The primary endpoint of this study was PFS. The secondary endpoints include time-to-treatment failure (TTF), response rate, OS, and incidences of adverse events ≥ grade 3.
Data collection
The researchers at each hospital maintained individual records for each patient as source data, including a copy of medical records, informed consent, image data, laboratory data, and other records. All data were collected by the Nippon Medical School Data Center. The data center oversaw the data sharing process within the trial. Clinical data entry, central monitoring, and data management were performed. Interim analysis and auditing were not undertaken for the study.
Sample size and statistical analysis
The RECOURSE trial was referred to for calculating sample size [12]. In the study, the PFS was 2.0 months. With a threshold and expected PFS of 2.1 and 3.5 months, respectively, the simulation results indicated a sample size of 29 with α = 0.05 (both sides) for 90% power based on the One Arm Binomial test using the SWOG statistical tool. If the estimated dropout was 7–8% cases, a target sample size of 32 was estimated.
All patients receiving TAS-102 plus bevacizumab chemotherapy were subjected to analysis. After enrollment, ineligible patients were excluded from this study. Dose intensity was performed when all patients completed two cycles of treatment and the last treatment. Response rates with 95% confidence intervals (CIs) were calculated for all eligible patients. The Kaplan–Meier method was used to calculate PFS and OS, and univariate analyses were performed using the log-rank test. Correlations were analyzed using Spearman’s rank correlation coefficient.
Results
We enrolled 32 patients who were treated between June 2016 and August 2017. The clinical cutoff date for the safety and efficacy analyses was 30 September 2019. No patient was lost to follow-up. The median age was 67 years (range: 45–78); 37.5% were women. Among the 32 patients, 23 (71.9%) and 9 (28.1%) had ECOG-PS scores of 0 and 1, respectively. Eighteen patients (56%) had RAS mutations. Table 1 summarizes the baseline characteristics of patients in this study.
At data cutoff, 31 (97%) patients who received TAS-102 plus bevacizumab had discontinued treatment, mainly because of tumor progression (Table 2). The median number of treatment cycles was four (IQR 2–6). The median overall relative dose intensity for TAS-102 was 0∙85 (0·77–0·90), and the median overall relative dose intensity for bevacizumab was 0∙90 (0·77–0·91). Dose modification of TAS-102 was undertaken in 14 (43.8%) patients. The main reason for dose reduction was hematological toxicity. All patients had previously received bevacizumab, and 65.6% of patients received bevacizumab as both first- and second-line treatment. Fifteen patients (46.9%) underwent fourth-line treatment and three patients (9.4%) received fifth-line treatment.
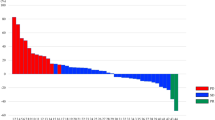
At the data cutoff, 29 (91%) patients had progressed or died. The median PFS was 4.5 months (95% CI 1.8–7.1), and the median OS was 9.2 months (95% CI 5.5–12.8) (Fig. 1). A waterfall plot of the changes in tumor size from baseline is shown in Fig. 2. Partial response was observed in two patients, and the disease control rate was 65.6%. The PFS of patients without RAS mutation was 5.4 months and that of patients with RAS mutation was 2.6 months (P = 0.16; log-rank test). The OS of patients without RAS mutation was 12.2 months and that of patients with RAS mutation was 9.3 months (P = 0.35; log-rank test).

Kaplan–Meier curves of a progression-free survival and b overall survival in all patients (n = 32). Progression-free survival and overall survival were 4.5 months and 9.2 months, respectively

Waterfall plot for changes in tumor size from baseline (red, PD; green, SD; blue, PR; yellow, CR)
Adverse events are shown in Table 3. The safety population included all patients who received chemotherapy. The most common adverse events above grade 3 were neutropenia (15 patients) followed by thrombocytopenia (4 patients). There were no non-hematologic adverse events of more than grade 4. No treatment-related deaths occurred.
Discussion
In this clinical study, we showed the survival benefit of TAS-102 plus bevacizumab in third-line treatment for CRC. This study met its primary endpoint PFS and confirmed the results of the C-TASK FORCE study. The C-TASK FORCE study included patients with second- and third-line treatment [5], and the Danish trial included patients who received up to sixth-line treatment [6]. Thus, this is the first trial of TAS-102 and bevacizumab combination therapy only in third-line treatment. This combination has the potential to be a therapeutic option for third-line treatment for metastatic CRC.
Previous studies showed that the PFS of patients with metastatic CRC with third-line treatment, including TAS-102 monotherapy, was approximately 2 months, and the OS was 6.7–7.8 months [12] [13] [14, 15]. The PFS in the C-TASK FORCE study, Danish trial and the present study was 3.7, 4.6 and 4.5 months, respectively. This study was performed as an exploratory study that included an examination of whether the median PFS was greater than 3.5 months and was not performed to determine statistical significance based on 95% confidence intervals. The OS in the C-TASK FORCE study and the present study was 11.4 and 9.2 months, respectively. The OS in the C-TASK FORCE study was better than that of our present study, because the C-TASK FORCE study included patients treated with second-line treatment. These results may indicate a survival benefit of TAS-102 and bevacizumab combination therapy.
Bevacizumab may have a survival benefit for patients with metastatic CRC as third-line treatment. The benefit of bevacizumab beyond PD (BBP) has been shown in the treatment of various cancers [16] [17]. The addition of bevacizumab to oxaliplatin-based chemotherapy reduces the frequency of splenic enlargement and the rate of thrombocytopenia [18]. In CRC, the survival benefit of BBP has only been shown in second-line treatment [19] [20] [21], and no clinical data were shown to clearly demonstrate the benefit of third-line BBP. In this study, all patients had previously received bevacizumab. Approximately 66% of patients received bevacizumab through the first- to third-line treatment. Becherirat et al. reported that bevacizumab treatment needs to be maintained, because discontinuous schedules tend to trigger tumor regrowth and increase tumor resistance and cancer stem cell heterogeneity [22]. Therefore, RAS mutant CRC may require continuous administration of anti-VEGF antibodies. Although the PFS of patients with RAS mutation was worse compared with patients with wild-type RAS, the OS of patients with RAS mutation was 9.3 months, indicating that BBP as third-line treatment may have survival benefits even for patients with RAS mutation. All 32 patients included in this study had been treated with bevacizumab: 21 patients received first- and second-line treatment, while 11 received only first- and second-line treatment. Future studies should compare the efficacy of the combination treatment in both patient groups.
This study included anti-EGFR antibody naïve patients with RAS wild-type tumors. This population has the therapeutic option of anti-EGFR antibodies with or without cytotoxic agents as third-line treatment. Although anti-EGFR antibodies are generally preferred, anti-VEGF antibodies seem to be an option for patients who cannot tolerate skin toxicity. Whether anti-VEGF antibodies should be continuously administered through first- to third-line treatment should be confirmed in further clinical trials.
Neutropenia is a serious issue in TAS-102 and bevacizumab combination therapy. The incidence of grade 3 or higher neutropenia in the present study, the CTASK FORCE study and the Danish trial was 46.9%, 72% and 67%, respectively [6]. Conversely, the incidence of grade 3 or higher neutropenia in the RECOURSE study and the TERRA study was 38% and 33.2%, respectively. Therefore, TAS-102 and bevacizumab may increase the risk of grade 3 or 4 neutropenia, possibly because of increased levels of FTD in hematopoietic cells. We previously reported that grade 3 or 4 neutropenia can be suppressed by changing the administration schedule of TAS-102 [10]. However, because this was a retrospective study, we are currently conducting a prospective study [23]. Although the administration schedule of TAS-102 is different, the dose intensity is the same, so neutropenia is predicted to be reduced.
The present study had some limitations. First, the study cohort was relatively small. However, the number of cases in the CTASK FORCE study was also small, with 25 patients. Second, this was a single-arm study and not a randomized trial. At present, there has been no trial of TAS-102 plus bevacizumab as third-line treatment only. Therefore, it seems possible to set the number of cases based on this result. Despite these limitations, the current results are valuable as data for third-line chemotherapy only. This study met its primary endpoint PFS, which is comparable to the results of the C-TASK FORCE study. The combination of TAS-102 plus bevacizumab has the potential to be an option for third-line therapy for metastatic CRC.
References
Bray F et al (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68(6):394–424
Nielsen DL et al (2014) A systematic review of salvage therapy to patients with metastatic colorectal cancer previously treated with fluorouracil, oxaliplatin and irinotecan+/− targeted therapy. Cancer Treat Rev 40(6):701–715
Emura T et al (2004) A novel antimetabolite, TAS-102 retains its effect on FU-related resistant cancer cells. Int J Mol Med 13(4):545–549
Chen J, Han M, Saif MW (2016) TAS-102 an emerging oral fluoropyrimidine. Anticancer Res 36(1):21–26
Kuboki Y et al (2017) TAS-102 plus bevacizumab for patients with metastatic colorectal cancer refractory to standard therapies (C-TASK FORCE): an investigator-initiated, open-label, single-arm, multicentre, phase 1/2 study. Lancet Oncol 18(9):1172–1181
Pfeiffer P et al (2020) TAS-102 with or without bevacizumab in patients with chemorefractory metastatic colorectal cancer: an investigator-initiated, open-label, randomised, phase 2 trial. Lancet Oncol S1470–2045(19):30827–30837
Kotani D et al (2019) Retrospective cohort study of trifluridine/tipiracil (TAS-102) plus bevacizumab versus trifluridine/tipiracil monotherapy for metastatic colorectal cancer. BMC cancer 19(1):1253–1253
Fujii H et al (2020) Bevacizumab in combination with TAS-102 improves clinical outcomes in patients with refractory metastatic colorectal cancer: a retrospective study. Oncologist 25(3):e469–e476. https://doi.org/10.1634/theoncologist.2019-0541
Matsuhashi N et al (2019) Combination chemotherapy with TAS-102 plus bevacizumab in salvage-line treatment of metastatic colorectal cancer: a single-center, retrospective study examining the prognostic value of the modified Glasgow prognostic score in salvage-line therapy of metastatic colorectal cancer. Mol Clin Oncol 11(4):390–396
Yoshida Y et al (2018) Biweekly administration of TAS-102 for neutropenia prevention in patients with colorectal cancer. Anticancer Res 38(7):4367–4373
Van Cutsem E et al (2016) ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol Off J Eur Soc Med Oncol 27(8):1386–1422
Mayer RJ et al (2015) Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med 372(20):1909–1919
Xu J et al (2018) Results of a randomized, double-blind, placebo-controlled, phase III trial of trifluridine/tipiracil (TAS-102) monotherapy in Asian patients with previously treated metastatic colorectal cancer: the TERRA study. J Clin Oncol 36(4):350–358. https://doi.org/10.1200/JCO.2017.74.3245
Grothey A et al (2013) Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381(9863):303–312
Loree JM, Kopetz S (2017) Recent developments in the treatment of metastatic colorectal cancer. Therap Adv Med Oncol 9(8):551–564
Takeda M et al (2016) Bevacizumab beyond disease progression after first-line treatment with bevacizumab plus chemotherapy in advanced nonsquamous non-small cell lung cancer (West Japan oncology group 5910L): an open-label, randomized, phase 2 trial. Cancer 122(7):1050–1059
Shoji T et al (2019) A New therapeutic strategy for recurrent ovarian cancer-bevacizumab beyond progressive disease. Healthcare (Basel) 7:3
Overman MJ et al (2018) The addition of bevacizumab to oxaliplatin-based chemotherapy: impact upon hepatic sinusoidal injury and thrombocytopenia. J Natl Cancer Inst 110(8):888–894
Grothey A et al (2008) Bevacizumab beyond first progression is associated with prolonged overall survival in metastatic colorectal cancer: results from a large observational cohort study (BRiTE). J Clin Oncol 26(33):5326–5334
Bennouna J et al (2013) Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. Lancet Oncol 14(1):29–37
Grothey A et al (2014) Bevacizumab exposure beyond first disease progression in patients with metastatic colorectal cancer: analyses of the ARIES observational cohort study. Pharmacoepidemiol Drug Saf 23(7):726–734
Becherirat S et al (2018) Discontinuous schedule of bevacizumab in colorectal cancer induces accelerated tumor growth and phenotypic changes. Trans Oncol 11(2):406–415
Yoshida Y et al (2019) A trial protocol of biweekly TAS-102 and bevacizumab as third-line chemotherapy for advanced/recurrent colorectal cancer: a phase II multicenter clinical trial (The TAS-CC4 study). J Anus Rect Colon 3(3):136–141
Acknowledgements
We thank all patients and co-workers for their participation and cooperation in the TAS-CC3 study. We also thank H. Nikki March, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Author information
Authors and Affiliations
Consortia
Contributions
Conception and design: YY, TY, KS, TO, and KK. Acquisition of data: YY, HK, SY, and HK. Analysis and interpretation of data: YY, TY, and TO. Writing, review, and/or revision of the manuscript: YY, TY, and SH. Administrative, technical, or material support: TY and AM. Study supervision: HY, HI, and KK. Other (recruitment of study subjects and oversight of study participants): YY, TY, HK, CK, KI, SY, HK, AF, HS, KY, and AM.
Corresponding author
Ethics declarations
Conflict of interest
Keiichiro Ishibashi has received research funding from Taiho and Chugai. Hideyuki Ishida has received research funding from Taiho and Chugai. Suguru Hasegawa has received research funding from Taiho and Takeda. The other authors declare that they have no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Yoshida, Y., Yamada, T., Kamiyama, H. et al. Combination of TAS-102 and bevacizumab as third-line treatment for metastatic colorectal cancer: TAS-CC3 study. Int J Clin Oncol 26, 111–117 (2021). https://doi.org/10.1007/s10147-020-01794-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10147-020-01794-8