
Abstract
Familial cerebral cavernous malformations (CCMs) occur with a frequency of 1 in 2000 and may cause recurrent headaches, seizures, and hemorrhagic stroke. Exon-scanning-based methods have identified intragenic mutations in three genes, CCM1, CCM2, and CCM3, in about 70% of familial CCM. To date, only two large CCM2 and a single large CCM3 deletion have been published. In addition to direct sequencing of all three CCM genes, we applied a newly developed multiplex ligation-dependent probe amplification gene dosage assay (MLPA) designed to detect genomic CCM1–3 deletions/duplications. Direct sequencing did not reveal a mutation in the index case who presented with multiple CCMs that had caused a generalized tonic-clonic seizure with Todd’s paralysis and headaches at the age of 5. In contrast, MLPA analyses detected a large deletion involving the entire CCM1 coding region in the proband and further affected members of this German CCM family. The MLPA results were corroborated by analyses of single nucleotide polymorphisms (SNPs) within the CCM1 gene. Thus, we here present the first report on a CCM1 gene deletion. Our results confirm a loss-of-function mutation mechanism for CCM1 and demonstrate that the use of MLPA enables a higher CCM mutation detection rate which is crucial for predictive testing of at-risk relatives.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Familial cerebral cavernous malformations (CCMs) (MIM 116860, 603284, 603285) are autosomal dominantly inherited vascular abnormalities with genetic heterogeneity and likely interaction among gene products [15]. Exon-by-exon screening approaches found CCM1 mutations in 43–54% of familial CCMs [2, 12]. Up to 22% were shown to carry a CCM2 mutation [4, 8] and less than 10% a CCM3 mutation [1, 5, 9, 12]. Two large CCM2 deletions [4] and one deletion involving the entire CCM3 gene [1] initially contributed to identification of the CCM2 and CCM3 genes via loss-of-heterozygosity mapping. Since large genomic deletions escape detection by conventional, nonquantitative polymerase chain reaction (PCR)-based mutation analysis, we adopted the multiplex ligation-dependent probe amplification (MLPA) gene dosage assay to screen for large deletions/duplications in the CCM1–3 genes. MLPA allows the relative quantitation of up to 50 different target DNA sequences in a single reaction and has been proven to be a reliable and sensitive method [7, 10]. We here present identification of a large, heterozygous deletion that encompasses the entire CCM1 coding region.
Patients and methods
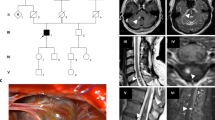
The index case is an 8-year-old boy (III-1, Fig. 1a) who experienced a generalized tonic-clonic seizure with Todd’s paralysis and headaches at the age of 5. Magnetic resonance imaging (MRI) of the brain showed multiple cavernous malformations, including a right temporomesial lesion (Fig. 1b,c). This symptomatic lesion with a diameter of 3.5 cm was completely excised microsurgically via a pterional approach. The postoperative course of the patient was uneventful. After 6 months, he did not require further antiepileptic medication.

a Pedigree of the German cerebral cavernous malformation (CCM) family (black circle = affected female, black square = affected male). b Axial T2-weighted magnetic resonance image (MRI) shows a large, right temporomesial cavernous malformation (white arrows) in the index case (arrow in a) before microsurgical excision. c Additional asymptomatic cavernous malformations (black arrows) were diagnosed in both cerebral hemispheres by gradient echo MRI. d Axial gradient echo MRI of the boy’s father shows two small asymptomatic cavernous malformations, left frontal and parietal (arrows). e One aunt (II-5 in a) also revealed an asymptomatic cavernous malformation in the left basal ganglia (arrow)
Family history revealed a paternal grandfather with multiple intracranial lesions and fatal hemorrhage at age 47. The patient’s father is clinically unaffected, but MRI revealed multiple small cavernous malformations which have not required surgical intervention so far (Fig. 1d). Neuroimaging of three further asymptomatic aunts (II-3, II-5, and II-7; Fig. 1a) revealed a small cavernous malformation in the basal ganglia of aunt II-5 (Fig. 1a,e).
Genetic testing was approved by the local ethics committees (University of Würzburg, Study 21/05; Philipps-University Marburg, Study 149/05). With informed consent, genomic DNA was extracted from peripheral blood lymphocytes. All coding CCM1–3 exons were directly sequenced on a Beckmann CEQ 8800 capillary electrophoresis system according to published protocols, with slight modifications [1, 2, 4]. Screening for large CCM alterations requires two MLPA kits (SALSA MLPA Kits P130 & P131 CCM; MRC Holland, Amsterdam, The Netherlands). The protocol provided by MRC Holland was followed without further optimization. CCM1–3 MLPA analyses of four control individuals in each test and all ten available family members were carried out using an ABI Prism 310 genetic analyzer. Haplotype analyses were performed for the index case and his parents using 19 intragenic single nucleotide polymorphisms (SNPs) (rs975707, 1064819, 1064820, 1064821, 11984192, 17164451, 2027950, 1034575, 10223994, 10282603, 10274699, 6953959, 12113704, 11542682, 1052043, 1063658, 1063659, 11542681, 1063660) and five polymorphic markers flanking the CCM1 locus (D7S2410, D7S1813, D7S2189, D7S646, and noninformative D7S689).
Results
Direct sequencing of all coding CCM1, CCM2, and CCM3 exons and adjacent splice sites did not reveal any pathological intragenic alterations in the index patient. In contrast, only the index case but none of the controls displayed a large deletion encompassing all CCM1 exons when tested by MLPA (Fig. 2b,e,f). CCM2 and CCM3 peaks and ratios did not differ between proband and controls (Fig. 2a,c,d). A second independent MLPA analysis included all ten family members of the second and third generation (Fig. 1a). The heterozygous CCM1 deletion was confirmed in the three affected family members only (data not shown). Thus, the CCM1 deletion was reproducible, segregates with the disease, and was not transmitted to children III-2 and III-3 and uncle II-6 (Fig. 1a), rendering neuroimaging unnecessary for these individuals and their offspring.

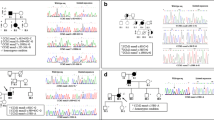
Multiplex ligation-dependent probe amplification gene dosage assay (MLPA) data demonstrating a heterozygous deletion of the entire CCM1 gene in the index case. Example of: a normal, and b pathological MLPA raw data (SALSA MLPA Kit P130). The electropherograms show reduced peaks for all CCM1 exons in the proband (asterisks), whereas CCM2 peaks (dots) are comparable between control and patient. Peaks from internal controls are not highlighted. e, f Quantitative analyses demonstrate that the relative peak areas are decreased to approximately 50% in the patient’s two noncoding and 15 coding CCM1 exons analyzed (black) when compared with internal (white) and c, d external controls and to e CCM2 (grey) and f CCM3 (grey) probes. Numbers below the columns in c, d indicate the CCM1–3 exons analyzed according to the manufacturer’s protocol
To further confirm the deletion detected by MLPA, haplotype analyses were performed with 19 intragenic CCM1 SNPs, none of which was found to be heterozygous in the patient and his affected father. Only three SNPs turned out to be informative. While the patient’s mother (II-2) is homozygous for G at rs975707, the father (II-1) is homo- or hemizygous for C (c.1-3078G > C). Since their son did not inherit a paternal C allele (Fig. 3), he is hemizygous for the maternal G allele. Similarly, the mother carries a homozygous C at rs2027950 and a homozygous T at rs6953959 (c.989 + 4389C > T), whereas the father’s sequence revealed a G (c.989 + 63C > G) and a C, respectively. The proband only shows the maternal C and T alleles. On the basis of the order of microsatellite markers linked to the disease locus and intragenic SNPs as D7S2410-D7S1813-D7S2189-rs975707-rs2027950-rs6953959-D7S646, the proband and his mother share the haplotype 1-2-2-G-C-T-1. The son inherited the disease haplotype 2-3-3-del-del-del-2 from his father, and this haplotype clearly lacks a second allele for three intragenic CCM1 SNPs (Fig. 3).

Haplotype analysis of the proband (III-1), his father (II-1), and his mother (II-2). While heterozygosity (i.e., the existence of two different alleles, a maternal and a paternal) for microsatellite markers flanking the CCM1 locus (D7S2410, D7S1813, D7S2189, D7S646) could be demonstrated for the index patient and his father, the proband carried only the maternal intragenic single nucleotide polymorphism (SNP) haplotype G-C-T, indicating hemizygosity (presence of only one copy of the respective DNA sequence) at the CCM1 locus for both father and son. The haplotype associated with the cerebral cavernous malformation (CCM) phenotype was determined as 2-3-3-del-del-del-2 (boxed) and lacks a second allele for three informative SNPs within the CCM1 gene (rs975707, rs2027950, rs6953959) (del = deletion), thus confirming the Multiplex ligation-dependent probe amplification gene dosage assay (MLPA) results
Discussion
The CCM1 deletion was found in a total of five CCM families in which four novel intragenic CCM mutations had been identified by direct sequencing ([11] and unpublished data). Based on microsatellite genotyping and cDNA analyses, previous publications reported that two out of ten CCM2 mutations [4] and one out of eight CCM3 mutations [1] were large deletions. An additional CCM3 mutation was described as possibly being due to a deletion of the genomic region that encompasses exon 5 [1]. Furthermore, a genomic deletion involving the 3’ end of CCM1 exon 18 and part of intron 18 was detected [6]. We anticipate that a significant proportion of CCM patients will display large deletions or duplications that remain undetected by direct sequencing of genomic DNA.
MLPA is a suitable and efficient method for identifying such CCM gene alterations. MLPA has been reported to be more precise, accurate, and time effective than real-time PCR [3]. Furthermore, deletions larger than the entire coding region, such as the CCM1 deletion presented in this report, would escape detection by RNA-based reverse transcriptase (RT)-PCR analysis using primers from the coding region, as has been described for deletions of the NF1 gene causing neurofibromatosis type 1 [14]. Fluorescence in situ hybridization (FISH) of chromosomes is much more laborious than is MLPA and would not be sensitive enough to detect single- or multiexon deletions/duplications. However, if sequencing of genomic DNA and MLPA fail to detect a mutation, RNA-based analysis is a complementary method that allows detection of splice mutations caused by, e.g., point mutations that activate a cryptic splice motif [13] or alterations located deep within a large intron. Thus, the application of MLPA and, in some instances RT-PCR, in addition to sequencing of genomic DNA is important for improving the mutation detection rate in CCM patients which is the basis for predictive testing of at-risk relatives.
References
Bergametti F, Denier C, Labauge P, Arnoult M, Boetto S, Clanet M, Coubes P, Echenne B, Ibrahim R, Irthum B, Jacquet G, Lonjon M, Moreau JJ, Neau JP, Parker F, Tremoulet M, Tournier-Lasserve E (2005) Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet 76:42–51
Cavé-Riant F, Denier C, Labauge P, Cecillon M, Maciazek J, Joutel A, Laberge-Le Couteulx S, Tournier-Lasserve E (2002) Spectrum and expression analysis of KRIT1 mutations in 121 consecutive and unrelated patients with Cerebral Cavernous Malformations. Eur J Hum Genet 10:733–740
Damgaard D, Nissen PH, Jensen LG, Nielsen GG, Stenderup A, Larsen ML, Faergeman O (2005) Detection of large deletions in the LDL receptor gene with quantitative PCR methods. BMC Med Genet 6:15
Denier C, Goutagny S, Labauge P, Krivosic V, Arnoult M, Cousin A, Benabid AL, Comoy J, Frerebeau P, Gilbert B, Houtteville JP, Jan M, Lapierre F, Loiseau H, Menei P, Mercier P, Moreau JJ, Nivelon-Chevallier A, Parker F, Redondo AM, Scarabin JM, Tremoulet M, Zerah M, Maciazek J, Tournier-Lasserve E (2004) Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am J Hum Genet 74:326–337
Guclu B, Ozturk AK, Pricola KL, Bilguvar K, Shin D, O’Roak BJ, Gunel M (2005) Mutations in apoptosis-related gene, PDCD10, cause cerebral cavernous malformation 3. Neurosurgery 57:1008–1013
Laberge-le Couteulx S, Jung HH, Labauge P, Houtteville JP, Lescoat C, Cecillon M, Marechal E, Joutel A, Bach JF, Tournier-Lasserve E (1999) Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet 23:189–193
Lalic T, Vossen RH, Coffa J, Schouten JP, Guc-Scekic M, Radivojevic D, Djurisic M, Breuning MH, White SJ, den Dunnen JT (2005) Deletion and duplication screening in the DMD gene using MLPA. Eur J Hum Genet 13:1231–1234
Liquori CL, Berg MJ, Siegel AM, Huang E, Zawistowski JS, Stoffer T, Verlaan D, Balogun F, Hughes L, Leedom TP, Plummer NW, Cannella M, Maglione V, Squitieri F, Johnson EW, Rouleau GA, Ptacek L, Marchuk DA (2003) Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet 73:1459–1464
Liquori CL, Berg MJ, Squitieri F, Ottenbacher M, Sorlie M, Leedom TP, Cannella M, Maglione V, Ptacek L, Johnson EW, Marchuk DA (2006) Low frequency of PDCD10 mutations in a panel of CCM3 probands: potential for a fourth CCM locus. Hum Mutat 27:118
Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G (2002) Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 30:e57
Sürücü O, Sure U, Gaetzner S, Stahl S, Benes L, Bertalanffy H, Felbor U (2006) Clinical impact of CCM mutation detection in familial cavernous angioma. Childs Nerv Syst 22:1461–1464
Verlaan DJ, Roussel J, Laurent SB, Elger CE, Siegel AM, Rouleau GA (2005) CCM3 mutations are uncommon in cerebral cavernous malformations. Neurology 65:1982–1983
Verlaan DJ, Siegel AM, Rouleau GA (2002) Krit1 missense mutations lead to splicing errors in cerebral cavernous malformation. Am J Hum Genet 70:1564–1567
Wimmer K, Yao S, Claes K, Kehrer-Sawatzki H, Tinschert S, De Raedt T, Legius E, Callens T, Beiglbock H, Maertens O, Messiaen L (2006) Spectrum of single- and multiexon NF1 copy number changes in a cohort of 1,100 unselected NF1 patients. Genes Chromosomes Cancer 45:265–276
Zawistowski JS, Stalheim L, Uhlik MT, Abell AN, Ancrile BB, Johnson GL, Marchuk DA (2005) CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Hum Mol Genet 14:2521–2531
Acknowledgements
The authors thank the patients and their family for their cooperation and MRC Holland for developing the MLPA kits. Ute Felbor receives an Emmy Noether-grant from the Deutsche Forschungsgemeinschaft (Fe 432/6-5) and Sonja Stahl a stipend from the Graduiertenkolleg 1048.
Author information
Authors and Affiliations
Corresponding author
Additional information
Comments
Comment for Publication NSR-08-06-0117.R1
Hildegard Kehrer-Sawatzki, Ulm, Germany
The results reported by Gaetzner et al. are important in the context of mutation screening in families or sporadic cases suffering from recurrent headaches, seizures, and hemorrhagic stroke attributable to cerebral cavernous malformations. These lesions are frequently caused by mutations in one of the three CCM genes, CCM1, CCM2, or CCM3. Gaetzner et al. successfully applied the MLPA technique and identified a deletion of the CCM1 gene in a family with several affected members. Since smaller and larger deletions are often difficult or even impossible to identify by the analysis of polymorphic markers, the MLPA technique applied in this study proved to be an efficient method to identify such alterations unambiguously. Thus, the MLPA technique is an important addition to the current mutation detection protocols if sequencing of exons failed to reveal mutations. The manuscript is written very well and the results are presented in a clear and illustrative manner.
Comments
Comm f Publication NSR-08-06-0117.R1
Hidetoshi Kasuya, Tokyo, Japan
The exact mechanism of pathogenesis for familial cavernous malformation is not known. Mutations at three loci (CCM1, CCM2, and CCM3) have been shown in familial cavernous malformation. Three CCM genes likely act through the same molecular pathway because familial cavernous malformations caused by different gene mutations are pathologically and phenotypically indistinguishable. There is growing evidence that CCM1 may play a role in regulating ß1 integrin-mediated angiogenesis through this product, which is involved with a bidirectional signaling pathway between the extracellular matrix and the cytoskeleton that uses an integrin-mediated cascade [1]. Many mutations have been reported in CCM1: frameshifts, nonsense mutations, changes in the invariant splice junctions, missense mutations, and 84-base pair deletion [2]. MLPA allows the detection of midsize alterations by simultaneously screening for the loss or duplication of up to 50 target sequences. By using this newly developed technique, Gaetzner et al. successfully present the first report on a CCM1 gene deletion larger than the entire coding region, which would escape detection by the techniques reported before. 1. Dashti SR, Hoffer A, Hu YC, Selman WR (2006) Molecular genetics of familial cerebral cavernous malformations. Neurosurg Focus 21(1):E2 2. Verlaan DJ, Davenport WJ, Stefan H, Sure U, Siegel AM, Rouleau GA (2002) Cerebral cavernous malformations: Mutations in Krit1. Neurology 58: 853-8574
Comments
Comment to NSR-08-06-0117.R1
CCM1 gene deletion identified by MLPA in cerebral cavernous malformation
Dietmar Krex, Dresden, Germany
The pathogenesis of familial cerebral cavernous malformation is based on genetic variants within three genes, CCM1, -2, and -3, which is in contrast to most other cerebral vascular malformations where disease-causing genes still have to be determined. Therefore, predictive testing of at-risk relatives is possible by the analysis of blood samples; a goal that has to be achieved, for instance, for arterio-venous malformations or intracranial aneurysms.
However, valid diagnostics are hampered, as genetic variants within CCM genes not only comprise various mutations but also large genomic deletions, which might lead to false negative results by standard sequencing techniques. Gaetzner et al. show that, by using the multiplex ligation-dependent probe amplification gene dosage assay (MLPA), this particular shortcoming of missing genomic deletions can be overcome, making the analysis more accurate. As MLPA is an established technique based on commercially available kits, it can be widely and easily used for improving the predictive value of genetic testing. We are looking forward to results from larger cohorts tested in that comprehensive manner.
Rights and permissions
About this article
Cite this article
Gaetzner, S., Stahl, S., Sürücü, O. et al. CCM1 gene deletion identified by MLPA in cerebral cavernous malformation. Neurosurg Rev 30, 155–160 (2007). https://doi.org/10.1007/s10143-006-0057-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10143-006-0057-1