
Abstract
The razor clam (Sinonovacula constricta) is an important aquaculture species, for which a high-density genetic linkage map would play an important role in marker-assisted selection (MAS). In this study, we constructed a high-density genetic map and detected quantitative trait loci (QTLs) for Sinonovacula constricta with an F1 cross population by using the specific locus amplified fragment sequencing (SLAF-seq) method. A total of 315,553 SLAF markers out of 467.71 Mreads were developed. The final linkage map was composed of 7516 SLAFs (156.60-fold in the parents and 20.80-fold in each F1 population on average). The total distance of the linkage map was 2383.85 cM, covering 19 linkage groups with an average inter-marker distance of 0.32 cM. The proportion of gaps less than 5.0 cM was on average 96.90%. A total of 16 suggestive QTLs for five growth-related traits (five QTLs for shell height, six QTLs for shell length, three QTLs for shell width, one QTL for total body weight, and one QTL for soft body weight) were identified. These QTLs were distributed on five linkage groups, and the regions showed overlapping on LG9 and LG13. In conclusion, the high-density genetic map and QTLs for S. constricta provide a valuable genetic resource and a basis for MAS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The razor clam (Sinonovacula constricta) is distributed in the intertidal, coastal, and estuarine waters of China, Japan, Korea, and Vietnam. The species is one of the four most valuable and popular edible clams in China, where it has been cultured for more than 100 years; however, there is a lack of improved parent clams for S. constricta.
Currently, marker-assisted selection (MAS) is considered an efficient and widely implemented method for selective breeding. A high-resolution genetic linkage map is essential for MAS in aquaculture because it is an indispensable tool for genome-wide quantitative trait locus (QTL) mapping. Construction of a genetic linkage map is based on the recombination frequencies between markers during the meiotic crossover of homologous chromosomes. The map distances indicate the physical distances between genes on the same chromosome. Genetic markers used for linkage map construction usually include restriction fragment length polymorphisms, random amplified polymorphic DNA, amplified fragment length polymorphisms (AFLP), simple sequence repeats, and single-nucleotide polymorphisms (SNPs) (Xu et al. 2015). Since the 1990s, genetic linkage maps have been constructed for more than 40 species of fish and shellfish, including second-generation linkage maps for several species (Yue 2014). With the development of next-generation sequencing (NGS) technologies, SNPs are preferred for constructing saturated maps because of advantages such as abundance, stability, simplicity, and a possibly direct relationship with phenotype (Lindblad-Toh et al. 2000), especially for species lacking genomic resources. Several methods have been used to identify SNP markers, for example restriction site-associated sequencing (RAD-seq) (Miller et al. 2007), double-digest RAD-seq (Peterson et al. 2012), and two-enzyme genotyping-by-sequencing (GBS) (Poland et al. 2012). Furthermore, specific locus amplified fragment sequencing (SLAF-seq) was developed as a new high-resolution strategy (Sun et al. 2013b); for high-throughput sequencing, a large number of polymorphic SLAF tags are obtained, and specific SNP sites are found on the basis of these SLAF tags. This approach has been successfully used for high-density genetic map construction in many species, regardless of the reference genome sequence. The efficiency of this method has been widely tested in plants such as soybean (Qi et al. 2014), cucumber (Xu et al. 2015), danshen (Tian et al. 2016), cauliflower (Zhao et al. 2016), and sorghum (Ji et al. 2017). In aquatic animals, high-density maps have been successfully constructed using SLAF-seq for the common carp (Cyprinus carpio) (Laghari et al. 2013), Pacific white shrimp (Litopenaeus vannamei) (Yu et al. 2015), and pearl mussel (Hyriopsis cumingii) (Bai et al. 2016).
Most economic traits are quantitative in nature and determined by many genes described as QTLs. QTL analysis based on linkage mapping reveals the genetic basis and inheritance patterns of these important traits (Li et al. 2012). QTL mapping has been performed using well-established procedures for correlating genetic and phenotypic variations in several species of fish (Abdelrahman et al. 2017), for instance resistance disease in Asian seabass (Liu et al. 2016) and gilthead seabream (Negrínbáez et al. 2016), growth traits in Japanese flounder (Cui et al. 2015) and kelp grouper (Kessuwan et al. 2016), and response to crowding in rainbow trout (Liu et al. 2015). In shellfish, the high fecundity and polymorphism levels may facilitate QTL mapping (Zhan et al. 2009). Recently, QTL analysis has conducted in bivalves, for example growth-related traits in scallop (Li et al. 2012; Petersen et al. 2012; Jiao et al. 2014) and oyster (Wang et al. 2016), shell color in scallop (Petersen et al. 2012), pearl-quality traits in the triangle pearl mussel (Bai et al. 2016) and pearl oysters (Jones et al. 2014), and disease resistance in the Pacific oyster (Sauvage et al. 2010).
To date, there is limited sequence information and markers (Niu et al. 2008; Niu et al. 2013; Wang et al. 2013; Niu et al. 2016) and no linkage map and QTLs for S. constricta. The aim of this study was to construct a high-density linkage map and detect growth-related QTLs by using SLAF-seq for S. constricta. The results would play a key role in MAS for the genetic improvement of S. constricta in the future.
Materials and Methods
Mapping Family and DNA Extraction
The mapping family was established in a hatchery in Sanmen County, Taizhou City, Zhejiang Province, China, in August 2013. The broodstock clams placed in baskets were spawned with water stimulation and continuous aeration in a breeding pool. The spawning clams were removed immediately in beakers of sand-filtered seawater. The sex was identified by observing the morphology of the sperm and eggs. Then, eggs and sperm from every two clams were mixed for artificial fertilization. The mating of the two parents generated an F1 cross. Briefly, fertilized eggs of each family were raised separately in 60-L barrels filled with sand-filtered seawater (salinity, 13‰) for hatching, feeding some sepecies of algae, such as Isochrysis galbana, Chaeroeeros moelleri, and Platymonas subcordiformis. After 3 months, the offspring each family were transferred to an outdoor nursery pond with nets to separate them.
At 10 months post-hatching, a total of 200 progeny were randomly collected from one family. The three traits including shell length (SL), shell height (SH), and shell width (SW) were measured by vernier calipe. The other two traits including soft body weight (SW) and total body weight (TW) were by electronic balance. Mantle tissues from the two parents and their progeny were dissected and stored in 75% ethanol at −20 °C. Genomic DNA was extracted using the phenol and chloroform method. After quantification using the NanoDrop-1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA) and integrity examination using agarose gel electrophoresis, the DNA samples were stored at −20 °C.
SLAF Library Preparation and High-Throughput Sequencing
A total of 153 individuals, including the two parents and 151 progeny, were de novo genotyped using SLAF-seq (Sun et al. 2013b). A pre-designed experiment was conducted to evaluate the enzymes and restriction fragment sizes. The reference was the genome of oyster (http://gigadb.org/dataset/100030). On the basis of the results of the experiment, an optimum scheme was confirmed and used to construct the SLAF library. Two enzymes, HaeIII and EcoRV-HF®, were used to digest the genomic DNA of all the samples. Then, a single nucleotide (A) overhang was added to the digested fragments by using the Klenow fragment (3′ → 5′ exon) and dATP at 37 °C. Duplex tag-labeled sequencing adapters (PAGE-purified, Life Technologies, USA) were then ligated to the A-tailed fragments using T4 DNA ligase. PCR was performed in reaction solutions containing the diluted restriction/ligation samples, dNTP, High-Fidelity DNA polymerase (NEB, Beverly, MA, USA), and PCR primers 5′-AATGATACGGCGACCACCGA-3′ (forward primer) and 5′-CAAGCAGAAG ACGGCATACG-3′ (reverse primer) (PAGE-purified). The PCR products were then purified using AgencourtAMPure XP beads (Beckman Coulter, High Wycombe, UK) and pooled. The fragments ranged from 264 to 364 base pairs and were gel-purified and diluted for pair-end sequencing on an Illumina High-seq 2500 sequencing platform. The ratio of raw high-quality reads with quality scores greater than Q30 (a quality score of 30 indicates a 0.1% chance of obtaining an error and thus 99.9% confidence) and guanine-cytosine (GC) content were calculated for quality control.
SLAF-seq Data Analysis and Genotyping
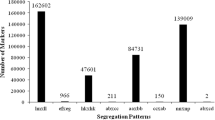
SLAF marker identification and genotyping were performed according to the procedures described by Sun et al. (2013b). Briefly, low-quality reads were filtered out and clean reads were clustered by similarity above 90% detected using one-to-one alignment with BLAT. Sequences clustered together were defined as one SLAF locus. SNP loci of each SLAF locus were then detected between parents, and SLAFs with more than three SNPs were filtered out first. Since S. constricta is a diploid species, SLAF loci with more than four alleles were defined as repetitive SLAFs and discarded subsequently. Only SLAFs with two to four alleles were identified as polymorphic and considered potential markers. Polymorphic SLAF markers were classified into eight segregation patterns (aa × bb, ab × cc, ab × cd, cc × ab, ef × eg, hk × hk, lm × ll, and nn × np). Since the F1 population of S. constricta is considered as a cross-pollinator (CP) population, only the SLAF markers with segregating patterns of ab × cd, ef × eg, hk × hk, lm × ll, nn × np, ab × cc, and cc × ab were used for the high-density genetic map construction. Finally, SLAFs with more than 80% integrity and more than 10-fold average sequence depths in the parents were used for the map construction.
Linkage map Construction and QTL Analysis
For linkage analysis, the sequencing data of the successful SLAF makers were used. The chi-square test (χ2) was performed to test the deviation of the polymorphic markers from the Mendelian inheritance ratios. Markers showing significant segregation distortion (P < 0.01) were filtered in the map.
To ensure a high-density and high-quality genetic map, a newly developed High Map strategy was used, which consists of four modules including linkage grouping, marker ordering, error genotyping correction, and map evaluation (Liu et al. 2014). The grouping module used the single-linkage clustering algorithm to cluster the markers into linkage groups, using a pairwise modified independence LOD (MLOD) score as distance metric, which less than four were filtered. Then, enhanced gibbs sampling, spatial sampling, and simulated annealing algorithms were combined to conduct an iterative process of marker ordering (Jansen et al. 2001; Van Ooijen 2011). The SMOOTH algorithm (van Os et al. 2005) was used to correct genotyping errors, and a k-nearest neighbor algorithm was applied to impute missing genotypes (Huang et al. 2012) following marker ordering. Map distances were estimated using the Kosambi mapping function (Kosambi 1943). Heat maps and haplotype maps were constructed to evaluate map quality.
MapQTL 5.0 software was used for QTL mapping with the interval mapping method. The LOD score threshold was set at 3.0 for QTL declaration, and QTLs that exceeded the LOD threshold were considered as suggestive QTLs. The LOD score threshold was determined using the 1000-permutation test with a confidence of 0.99. QTLs with LOD scores greater than the threshold at a confidence of 0.99 were declared significant.
Results
Analysis of SLAF-seq Data
The control sequencing data were evaluated to ensure the validity of the SLAF library construction. Two restriction endonucleases (HaeIII and EcoRV-HF) were used for the SLAF library construction. For the control, the percentage of paired-end mapping reads was 85.97%, and the percentage of digestion was 94.75%.
In this study, high-throughput sequencing of the SLAF library generated 93.09 Gb of data containing 467.71 Mreads with a length of 100 bp. The Q30 percentage was 90.22%, and the GC content was 39.99%. The number of reads in the male and female parents was 19,793,475 and 15,922,216, respectively. On average, 2,860,862 reads were generated in the F1 mapping population (Table S1).
SLAF Marker Detection and Genotype Definition
In the male parent, the number of SLAFs was 218,251, and the average depth of each SLAF marker was 42.70-fold. In the female, 224,896 SLAFs were generated, with an average depth of 30.71-fold for each SLAF. Then, the abnormal SLAF markerslabes, which were detected at a higher depth in the offspring but not in the parents, were tested for paternity. The individuals with more than 0.3% abnormal markers were identified as abnormal paternity individuals. Excluding 34 abnormal individuals, an analysis of the 117 progeny from the F1 mapping population indicated that 176,706 SLAFs were generated, with an average depth of 6.70-fold for each offspring (Table 1).
Of the 315,553 high-quality SLAFs, 144,920 were polymorphic with a polymorphism percentage of 45.93%. After filtering out the SLAFs lacking parent information, 96,655 SLAFs were classified into eight segregation patterns. The segregation patterns were as follows: ab × cd, ef × eg, hk × hk, lm × ll, nn × np, aa × bb, ab × cc, and cc × ab (Fig. S1). For mapping with the F1 population, seven segregation patterns (ab × cd, ef × eg, hk × hk, lm × ll, nn × np, ab × cc, and cc × ab) were used for the genetic map construction. To further improve SLAF accuracy, the SLAFs with more than 80% integrity and more than 10-fold average sequence depths in the parents were used for the map construction. Finally, 8212 of the 96,655 markers were defined as effective and selected for the construction of the linkage map. The segregation types for these effective markers are shown in Table 2.
Construction of the Genetic Map
After completing the data preparation, 7516 of the 8212 SLAFs (91.52%) containing 721 segregation distortion markers, with 185.39-fold sequence depth in the male parent, 127.81-fold in the female parent, and 20.80-fold in each F1 population on average, were used for constructing the map (Table 3). The final map included 7516 markers on 19 linkage groups (LGs), which is consistent with the diploid chromosome number of the razor clam (2 N = 38) (Fig. 1), 2383.85 cM in length, and an average inter-marker distance of 0.32 cM. The number of markers mapped on each linkage group varied from 104 markers in LG5 to 716 markers in LG18, with an average of 395 markers per LG. The largest LG was LG18 covering a genetic length of 180.98 cM, and the smallest LG was LG17 containing only 180 markers with a length of 70.66 cM. The value of gap less than 5 cM on the 19 LGs ranged from 88.82 to 99.70% (average, 96.90%). The maximum gap was 20.92 cM, which was observed in LG13. The largest average inter-marker distance was 0.93 cM in LG5. The two groups (LG11 and LG15) were the most saturated with an average marker density of 0.23 cM (Table 4).

The diagram of integrated linkage map for S. constricta and the shared markers between female and male maps with red color
In this study, sex-specific maps were also constructed (Table S2, S3). The female map contained 4180 markers covering 2731.31 cM (Table S4), while the male map contained 4189 markers spanning 1815.64 cM (Table S5). The average inter-marker distances for the male and female maps were 0.44 and 0.66 cM, respectively.
QTL Analysis for Growth Traits
The basic statistics for the growth traits are listed in Table S6. Pairwise comparisons among the five growth traits using Pearson’s correlation revealed that all of them were highly correlated with correlation coefficients ranging from 0.803 to 0.979 (P < 0.01; Table S7). On the basis of the high-density genetic map, a total of 16 QTLs were detected on LG9, LG11, LG13, LG18, and LG19 (Table 5). The proportion of phenotypic variance explained by a single QTL (r2) ranged from 11.33 to 15.44% and LOD scores, from 3.04 to 3.77. Five QTLs for shell height are located on LG9 (79.002–79.404, 86.079–86.539, and 88.654–98.322 cM) and LG13 (94.737–95.599 and 104.344 cM). For shell length, one QTL was detected at 68.871–70.03 cM on LG18 and five QTLs on LG19 containing 79.163–79.839, 90.979–92.28, 98.383, 113.354, and 124.377–125.239 cM. Three QTLs for shell width are located on the regions containing 44.615–45.931 and 47.232 cM on LG11 and 104.344 cM on LG13. The same QTL for total body weight and soft body weight is 92.308 cM on LG9, showing a percentage of phenotypic variance explained (PVE) of 13.6 and 13.1%, respectively.
Discussion
SLAF-seq is a highly automated technique performed by sequencing the paired ends of the sequence-specific restriction fragment length on the basis of high-throughput sequencing. In this study, SLAF-seq was used to identify a set of SLAF markers in the razor clam, S. constricta. We constructed a SLAF library and obtained 93.09-Gb data containing 467,71 Mreads. Subsequently, 315,553 SLAF markers were detected, including 68,507 high-quality SLAF markers with a polymorphism rate of 21.71%, and a total of 8212 polymorphic markers were identified as effective for linkage mapping. Moreover, the average sequencing depths were 185.39-fold and 127.81-fold for the male and female parents, respectively, and an average 20.87-fold for each progeny; this provided a high level of genotyping accuracy. The genotype integrity of these markers was more than 80% in the mapping population, and the average integrity was 99.95%, confirming the higher integrity and accuracy. Our results showed that SLAF-seq is a powerful method for marker identification and high-density linkage map construction.
Genetic linkage maps have been constructed in a few bivalve species. However, traditionally, AFLP and microsatellite markers were selected for linkage analyses in organisms lacking enough genomic information, such as the Pacific Oyster (Crassostrea gigas) (Hubert and Hedgecock 2004), bay scallop (Argopecten irradians) (Qin et al. 2007), European flat oyster (Ostrea edulis) (Lallias et al. 2007), pearl oyster (Pinctada martensii) (Shi et al. 2009), Pacific lion-paw scallop (Nodipecten subnodosus) (Petersen et al. 2012), and freshwater pearl mussel (H. cumingii) (Bai et al. 2015). These maps were mostly constructed using several hundred molecular markers. In this study, we constructed the first genetic linkage map for S. constricta by using SLAF-seq. The map consists of 7516 SLAF markers and spans 2383.85 cM, with an average distance of 0.32 cM. The density of the current linkage map is much higher than that of the maps constructed using GBS for bivalves such as Chlamys farreri (3806-2b-RAD markers, 0.41 cM) (Jiao et al. 2014), Pinctada fucata (3117 2b–RAD markers, 0.39 cM) (Shi et al. 2014), and H. cumingii (4983 SLAF markers, 1.81 cM) (Bai et al. 2016).
The number of SLAF markers on each LG was different; the markers were not randomly distributed, with some clear marker-dense regions and some marker deserts (Ji et al. 2017). In the map, an average percentage of 96.90% was obtained for the gaps on linkage groups less than 5 cM. Although the average distance between adjacent markers on the map was short (only 0.32 cM), gaps larger than 10 cM on 11 linkage groups suggested that such gaps are not restricted to a particular group. This pattern may contribute to the non-random distribution of the markers and uneven marker polymorphism and recombination rates between the mapping parents (Wang et al. 2012; Sun et al. 2013a). The use of other enzymes for SLAF library preparation and increasing the size of the mapping population may improve the map in the future (Bai et al. 2016).
Sex-specific differences in recombination rates are not uncommon and have been reported in vertebrate and invertebrates. The males have substantially lower meiotic recombination rate than females, for organisms with a chromosomal mechanism of sex determination (Jones et al. 2013). This has been termed heterochiasmy and firstly described by Haldane (1922) and Huxley (1928) (‘Haldane–Huxley rule’) (Haldane 1922; Huxley 1928). In this study, results observed here for S. constricta show that the male map (1815.64 cM) is shorter than the female map (2731.31 cM), suggesting a significant female bias in recombination with an overall ratio of female-to-male recombination of 1.5:1. The phenomenon has been reported in pearl oyster (Jones et al. 2013), the Pacific oyster (Hubert and Hedgecock 2004; Li and Guo 2004), and other fishes (Sakamoto et al. 2000; Franch et al. 2006; Lien et al. 2011). However, the unusual pattern of sex-specific recombination rate is not well understood. Several theories have been proposed, such as differing environments in which the germ cells develop, temporal differences in initiation of meiosis between the sexes, and pairing and synapses of the homologs at meiosis differed between oocytes and spermatocytes (Wang et al. 2016). In addition, for species with no specialized sex chromosomes, there will be another underling phenomenon of the timing, duration, or biological features associated with meiosis that is responsible for the observed differences between the sexes (Jones et al. 2013).
Segregation distortion is a common phenomenon, indicating that the genotypic frequency deviates from a typical Mendelian ratio (Ji et al. 2017). Previous studies have shown that a large number of segregation distortions occur in plant (Ji et al. 2017), fish (Woram et al. 2004; Amores et al. 2014), shellfish (Qin et al. 2007; Wang et al. 2016), and crustacean (Qiu et al. 2016) species. In general, markers with segregation distortion frequently affect the accuracy of genetic maps (Tian et al. 2016). However, further studies have proven that the presence of segregation distortion markers will not affect the use of linkage maps in applications such as QTL mapping (Zhang et al. 2010). Moreover, the use of distorted markers for linkage map construction could increase the genome coverage of the genetic map and help improve the detection of linked QTLs (Xu 2008), and discarding them could potentially remove massive amounts of information and decrease genome coverage (Luo et al. 2005). Segregation distortion should be performed in a non-random and consistent distribution pattern. In this study, markers with significant segregation distortion (P < 0.01) were initially excluded to guarantee maximum map accuracy. A total of 721 markers (7.82%) that displayed significant distorted segregation (P < 0.05) were used for constructing the linkage map. These markers were distributed on 13 linkage groups, especially on LG1, LG2, LG15, LG16, LG17, and LG19. The uneven distribution of the distorted markers may suggest that marker distortion was not caused by technical limitations or other typing errors (Wang et al. 2016). In bivalves, a high genetic load of deleterious recessive genes was detected, resulting in strong zygotic selection during the larval stage (Launey and Hedgecock 2001); segregation distortion was reduced by genotyping 11-day-old outbred larvae (Hubert and Hedgecock 2004). However, the mechanism underlying segregation distortion is still unclear, and it is recognized as a potentially powerful evolutionary force (Taylor and Ingvarsson 2003). Biological causes such as gametic and zygotic selection, duplicated genes, non-homologous recombination, and non-homologous or translocation loci on chromosomes may be the main causes (Faure et al. 1993; Hubert and Hedgecock 2004; Hubert et al. 2010).
Growth is an important trait for the selective breeding of shellfish. A high-density linkage map is an effective tool for the fine mapping of QTLs for these important traits. QTLs for growth-related traits have been developed in bivalves such as oyster (Guo et al. 2012; Shi et al. 2014; Wang et al. 2016) and scallop (Li et al. 2012; Petersen et al. 2012; Jiao et al. 2014). However, to the best of our knowledge, there are no reports of QTLs for S. constricta. In this study, five QTLs for shell height, six QTLs for shell length, three QTLs for shell width, one QTL for total body weight, and one QTL for soft body weight were identified. Moreover, the QTL regions focus on primary linkage groups (such as LG9, LG11, LG13, and LG19), indicating that genes from different chromosomes may contribute to the same trait. Moreover, the relatively low PVE indicated that no major loci were detected and the growth-related traits were regulated by many genes with low effects (Wang et al. 2016). In the present study, the PVE explained by these QTLs ranged from 11.33 to 15.44%, which is higher than that of the oyster (average, 5.4%) (Wang et al. 2016). Similarly, in small abalone, a total of 15 suggestive QTL were identified based on a high-density genetic map with 3717 SNPs (Ren et al. 2016). However, a limited number of QTL for growth-related traits were detected due to genetic maps with low density. For instance, in Pacific oyster, three significant QTLs for growth-related traits were detected using 426 markers, which explained 0.6, 7.5, and 13.8% of the phenotypic variance respectively (Guo et al. 2012). Then, 27 QTLs for five growth-related traits were detected based on the second-generation linkage map of Pacific oyster with 3367 SNPs (Wang et al. 2016). It is suggested that the density of the genetic map is a key to perform QTL fine mapping of important growth-related traits.
Interestingly, overlaps were observed among the QTL regions for multiple traits. For example, the QTL regions for shell width and shell height are overlapped on LG13. In particular, not only was the same QTL detected for total body weight and soft body weight on LG9, but it also overlapped with the region for shell height on LG9. Thus, this marker (92.308 cM, LG9) may be an important genomic region involved in controlling the growth traits. The overlapping of QTLs has been reported in P. fucata (Shi et al. 2014), Haliotis diversicolor (Ren et al. 2016), and Lates calcarifer (Xia et al. 2014), suggesting that genes in that particular region have significant pleiotropic effects. Furthermore, Pearson’s analysis showed that the total body weight had the highest correlation with soft body weight (r = 0.979, P < 0.01). Besides, total body weight also had a significantly high correlation with the other traits. Selection for total body weight will have positive effects on other traits, indicating the best trait for the breeding of S. constricta. We would perform further studies to identify the genes located within the QTL regions.
References
Abdelrahman H, ElHady M, Alcivar-Warren A, Allen S, Al-Tobasei R, Bao L, Beck B, Blackburn H, Bosworth B, Buchanan J, Chappell J, Daniels W, Dong S, Dunham R, Durland E, Elaswad A, Gomez-Chiarri M, Gosh K, Guo X, Hackett P, Hanson T, Hedgecock D, Howard T, Holland L, Jackson M, Jin Y, Kahlil K, Kocher T, Leeds T, Li N, Lindsey L, Liu S, Liu Z, Martin K, Novriadi R, Odin R, Palti Y, Peatman E, Proestou D, Qin G, Reading B, Rexroad C, Roberts S, Salem M, Severin A, Shi H, Shoemaker C, Stiles S, Tan S, Tang KF, Thongda W, Tiersch T, Tomasso J, Prabowo WT, Vallejo R, van der Steen H, Vo K, Waldbieser G, Wang H, Wang X, Xiang J, Yang Y, Yant R, Yuan Z, Zeng Q, Zhou T (2017) Aquaculture genomics, genetics and breeding in the United States: current status, challenges, and priorities for future research. BMC Genomics 18:191
Amores A, Catchen J, Nanda I, Warren W, Walter R, Schartl M, Postlethwait JH (2014) A RAD-tag genetic map for the platyfish (Xiphophorus maculatus) reveals mechanisms of karyotype evolution among teleost fish. Genetics 197:625–641
Bai ZY, Han X, Luo M, Lin JY, Wang GL, Li JL (2015) Constructing a microsatellite-based linkage map and identifying QTL for pearl quality traits in triangle pearl mussel (Hyriopsis cumingii). Aquaculture 437:102–110
Bai ZY, Han XK, Liu XJ, Li QQ, Li JL (2016) Construction of a high-density genetic map and QTL mapping for pearl quality-related traits in Hyriopsis cumingii. Sci Rep 6:32608
Cui Y, Wang H, Qiu X, Liu H, Yang R (2015) Bayesian analysis for genetic architectures of body weights and morphological traits using distorted markers in Japanese flounder Paralichthys olivaceus. Mar Biotechnol 17:693–702
Faure S, Noyer JL, Horry JP, Bakry F, Lanaud C, Gonzalez de Leon D (1993) A molecular marker-based linkage map of diploid bananas (Musa acuminata). Theor Appl Genet 87:517–526
Franch R, Louro B, Tsalavouta M, Chatziplis D, Tsigenopoulos CS, Sarropoulou E, Antonello J, Magoulas A, Mylonas CC, Babbucci M, Patarnello T, Power DM, Kotoulas G, Bargelloni L (2006) A genetic linkage map of the hermaphrodite teleost fish Sparus aurata L. Genetics 174:851–861
Guo X, Li Q, Wang QZ, Kong LF (2012) Genetic mapping and QTL analysis of growth-related traits in the Pacific oyster. Mar Biotechnol (NY) 14:218–226
Haldane JBS (1922) Sex ratio and unisexual sterility in hybrid animals. J Genet 12:101–109
Huang XH, Zhao Y, Wei XH, Li CY, Wang A, Zhao Q, Li WJ, Guo YL, Deng LW, Zhu CR, Fan DL, Lu YQ, Weng QJ, Liu KY, Zhou TY, Jing YF, Si LZ, Dong GJ, Huang T, Lu TT, Feng Q, Qian Q, Li JY, Han B (2012) Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat Genet 44:32–U53
Hubert S, Hedgecock D (2004) Linkage maps of microsatellite DNA markers for the pacific oyster Crassostrea gigas. Genetics 168:351–362
Hubert S, Higgins B, Borza T, Bowman S (2010) Development of a SNP resource and a genetic linkage map for Atlantic cod (Gadus morhua). BMC Genomics 11:191
Huxley JS (1928) Sexual difference of linkage in Gammarus chevreuxi. J Genet 20:145–156
Jansen J, de Jong AG, van Ooijen JW (2001) Constructing dense genetic linkage maps. Theor Appl Genet 102:1113–1122
Ji GS, Zhang QJ, Du RH, Lv P, Ma X, Fan S, Li SY, Hou SL, Han YC, Liu GQ (2017) Construction of a high-density genetic map using specific-locus amplified fragments in sorghum. BMC Genomics 18:51
Jiao W, Fu X, Dou J, Li H, Su H, Mao J, Yu Q, Zhang L, Hu X, Huang X (2014) High-resolution linkage and quantitative trait locus mapping aided by genome survey sequencing: building up an integrative genomic framework for a bivalve mollusc. DNA Res 21:85–101
Jones DB, Jerry DR, Khatkar MS, Raadsma HW, Zenger KR (2013) A high-density SNP genetic linkage map for the silver-lipped pearl oyster, Pinctada maxima: a valuable resource for gene localisation and marker-assisted selection. BMC Genomics 14:810
Jones DB, Jerry DR, Khatkar MS, Moser G, Raadsma HW, Taylor JJ, Zenger KR (2014) Quantitative trait loci and genetic association analysis reveals insights into complex pearl quality traits in donor silver-lipped pearl oysters. Aquaculture 434:476–485
Kessuwan K, Kubota S, Liu Q, Sano M, Okamoto N, Sakamoto T, Yamashita H, Nakamura Y, Ozaki A (2016) Detection of growth-related quantitative trait loci and high-resolution genetic linkage maps using simple sequence repeat markers in the kelp grouper (Epinephelus bruneus). Mar Biotechnol 18:57–84
Kosambi DD (1943) The estimation of map distances from recombination values. Ann Hum Genet 12:172–175
Laghari MY, Lashari P, Zhang X, Xu P, Xin B, Zhang Y, Narejo NT, Sun X (2013) Mapping quantitative trait loci (QTL) for body weight, length and condition factor traits in backcross (BC1) family of common carp ( Cyprinus carpio L.) Mol Biol Rep 41:721–731
Lallias D, Beaumont AR, Haley CS, Boudry P, Heurtebise S, Lapegue S (2007) A first-generation genetic linkage map of the European flat oyster Ostrea edulis (L.) based on AFLP and microsatellite markers. Anim Genet 38:560–568
Launey S, Hedgecock D (2001) High genetic load in the Pacific oyster Crassostrea gigas. Genetics 159:255–265
Li L, Guo X (2004) AFLP-based genetic linkage maps of the pacific oyster Crassostrea gigas Thunberg. Mar Biotechnol (NY) 6:26–36
Li H, Liu X, Zhang G (2012) A consensus microsatellite-based linkage map for the hermaphroditic bay scallop (Argopecten irradians) and its application in size-related QTL analysis. PLoS One 7:e46926
Lien S, Gidskehaug L, Moen T, Hayes BJ, Berg PR, Davidson WS, Omholt SW, Kent MP (2011) A dense SNP-based linkage map for Atlantic salmon (Salmo salar) reveals extended chromosome homeologies and striking differences in sex-specific recombination patterns. BMC Genomics 12:615
Lindblad-Toh K, Winchester E, Daly MJ, Wang DG, Hirschhorn JN, Laviolette JP, Ardlie K, Reich DE, Robinson E, Sklar P, Shah N, Thomas D, Fan JB, Gingeras T, Warrington J, Patil N, Hudson TJ, Lander ES (2000) Large-scale discovery and genotyping of single-nucleotide polymorphisms in the mouse. Nat Genet 24:381–386
Liu DY, Ma CX, Hong WG, Huang L, Liu M, Liu H, Zeng HP, Deng DJ, Xin HG, Song J, Xu CH, Sun XW, Hou XL, Wang XW, Zheng HK (2014) Construction and analysis of high-density linkage map using high-throughput sequencing data. PLoS One 9:e98855
Liu S, Vallejo RL, Gao G, Palti Y, Weber GM, Hernandez A, Rd RC (2015) Identification of single-nucleotide polymorphism markers associated with cortisol response to crowding in rainbow trout. Mar Biotechnol 17:328
Liu P, Wang L, Wan ZY, Ye BQ, Huang S, Wong SM, Yue GH (2016) Mapping qtl for resistance against viral nervous necrosis disease in Asian seabass. Mar Biotechnol 18:107–116
Luo L, Zhang YM, Xu S (2005) A quantitative genetics model for viability selection. Heredity (Edinb) 94:347–355
Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA (2007) Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res 17:240–248
Negrínbáez D, Navarro A, Rodríguezramilo ST, Afonso JM, Zamorano MJ (2016) Identification of quantitative trait loci associated with the skeletal deformity lsk complex in gilthead seabream (Sparus aurata L.) Mar Biotechnol 18:98–106
Niu DH, Li JL, Liu DB (2008) Polymorphic microsatellite loci for population studies of the razor clam, Sinonovacula constricta. Conserv Genet 9:1393–1394
Niu D, Wang L, Sun F, Liu Z, Li J (2013) Development of molecular resources for an intertidal clam, Sinonovacula constricta, using 454 transcriptome sequencing. PLoS One 8:e67456
Niu DH, Wang F, Xie SM, Sun FY, Wang Z, Peng MX, Li JL (2016) Developmental transcriptome analysis and identification of genes involved in larval metamorphosis of the razor clam, Sinonovacula constricta. Mar Biotechnol 18:168–175
Petersen JL, Baerwald MR, Ibarra AM, May B (2012) A first-generation linkage map of the Pacific lion-paw scallop (Nodipecten subnodosus): initial evidence of QTL for size traits and markers linked to orange shell color. Aquaculture 350:200–209
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One 7:e37135
Poland JA, Brown PJ, Sorrells ME, Jannink JL (2012) Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS One 7:e32253
Qi ZM, Huang L, Zhu RS, Xin DW, Liu CY, Han X, Jiang HW, Hong WG, Hu GH, Zheng HK, Chen QS (2014) A high-density genetic map for soybean based on specific length amplified fragment sequencing. PLoS One 9:e104871
Qin Y, Liu X, Zhang H, Zhang G, Guo X (2007) Genetic mapping of size-related quantitative trait loci (QTL) in the bay scallop (Argopecten irradians) using AFLP and microsatellite markers. Aquaculture 272:281–290
Qiu G-F, Xiong L-W, Liu Z-Q, Yan Y-L, Shen H (2016) A first generation microsatellite-based linkage map of the Chinese mitten crab Eriocheir sinensis and its application in quantitative trait loci (QTL) detection. Aquaculture 451:223–231
Ren P, Peng W, You W, Huang Z, Guo Q, Chen N, He P, Ke J, Gwo J-C, Ke C (2016) Genetic mapping and quantitative trait loci analysis of growth-related traits in the small abalone Haliotis diversicolor using restriction-site-associated DNA sequencing. Aquaculture 454:163–170
Sakamoto T, Danzmann RG, Gharbi K, Howard P, Ozaki A, Khoo SK, Woram RA, Okamoto N, Ferguson MM, Holm LE, Guyomard R, Hoyheim B (2000) A microsatellite linkage map of rainbow trout (Oncorhynchus mykiss) characterized by large sex-specific differences in recombination rates. Genetics 155:1331–1345
Sauvage C, Boudry P, de Koning DJ, Haley CS, Heurtebise S, Lapegue S (2010) QTL for resistance to summer mortality and OsHV-1 load in the Pacific oyster (Crassostrea gigas). Anim Genet 41:390–399
Shi Y, Hong K, Guo X, Gu Z, Wang Y, Wang A (2009) Genetic linkage map of the pearl oyster, Pinctada martensii (Dunker). Aquac Res 41:35–44
Shi Y, Wang S, Gu Z, Lv J, Zhan X, Yu C, Bao Z, Wang A (2014) High-density single nucleotide polymorphisms linkage and quantitative trait locus mapping of the pearl oyster, Pinctada fucata martensii Dunker. Aquaculture 434:376–384
Sun L, Yang W, Zhang Q, Cheng T, Pan H, Xu Z, Zhang J, Chen C (2013a) Genome-wide characterization and linkage mapping of simple sequence repeats in mei (Prunus mume Sieb. Et Zucc.) PLoS One 8:e59562
Sun XW, Liu DY, Zhang XF, Li WB, Liu H, Hong WG, Jiang CB, Guan N, Ma CX, Zeng HP, Xu CH, Song J, Huang L, Wang CM, Shi JJ, Wang R, Zheng XH, Lu CY, Wang XW, Zheng HK (2013b) SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS One 8:e58700
Taylor DR, Ingvarsson PK (2003) Common features of segregation distortion in plants and animals. Genetica 117:27–35
Tian L, Guo L, Pan Y, Qi Z, Wang J, Song Z (2016) Construction of the first high-density genetic linkage map of Salvia miltiorrhiza using specific length amplified fragment (SLAF) sequencing. Sci Rep 6:24070
Van Ooijen JW (2011) Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet Res 93:343–349
van Os H, Stam P, Visser RGF, van Eck HJ (2005) SMOOTH: a statistical method for successful removal of genotyping errors from high-density genetic linkage data. Theor Appl Genet 112:187–194
Wang WX, Huang SM, Liu YM, Fang ZY, Yang LM, Hua W, Yuan SX, Liu SY, Sun JF, Zhuang M, Zhang YY, Zeng AS (2012) Construction and analysis of a high-density genetic linkage map in cabbage (Brassica oleracea L. var. capitata). BMC Genomics 13:523
Wang L, Niu D, Li J (2013) Charaterization of novel EST-derived SNP markers by using 454 pyrosequencing in Sinonovacula constricta. Conserv Genet Resour 5:191–193
Wang J, Li L, Zhang G (2016) A high-density snp genetic linkage map and qtl analysis of growth-related traits in a hybrid family of oysters (Crassostrea gigas × Crassostrea angulata) using genotyping-by-sequencing. G3-Genes Genomes Genetics 6: 1417-1426
Woram RA, Mcgowan C, Stout JA, Gharbi K, Ferguson MM, Hoyheim B, Davidson EA, Davidson WS, Rexroad C, Danzmann RG (2004) A genetic linkage map for Arctic char ( Salvelinus alpinus ): evidence for higher recombination rates and segregation distortion in hybrid versus pure strain mapping parents. Genome 47:304–315
Xia JH, Lin G, He X, Yunping B, Liu P, Liu F, Sun F, Tu R, Yue GH (2014) Mapping quantitative trait loci for omega-3 fatty acids in Asian seabass. Mar Biotechnol (NY) 16:1–9
Xu SZ (2008) Quantitative trait locus mapping can benefit from segregation distortion. Genetics 180:2201–2208
Xu XW, Xu RX, Zhu BY, Yu T, Qu WQ, Lu L, Xu Q, Qi XH, Chen XH (2015) A high-density genetic map of cucumber derived from specific length amplified fragment sequencing (SLAF-seq). Front Plant Sci 5:768
Yu Y, Zhang X, Yuan J, Li F, Chen X, Zhao Y, Huang L, Zheng H, Xiang J (2015) Genome survey and high-density genetic map construction provide genomic and genetic resources for the Pacific White Shrimp Litopenaeus vannamei. Sci Rep 5:15612
Yue GH (2014) Recent advances of genome mapping and marker-assisted selection in aquaculture. Fish Fish 15:376–396
Zhan A, Hu J, Hu X, Hui M, Wang M, Peng W, Huang X, Wang S, Lu W, Sun C, Bao Z (2009) Construction of microsatellite-based linkage maps and identification of size-related quantitative trait loci for Zhikong scallop (Chlamys farreri). Anim Genet 40:821–831
Zhang L, Wang S, Li H, Deng Q, Zheng A, Li S, Li P, Li Z, Wang J (2010) Effects of missing marker and segregation distortion on QTL mapping in F2 populations. Theor Appl Genet 121:1071–1082
Zhao Z, Gu H, Sheng X, Yu H, Wang J, Huang L, Wang D (2016) Genome-wide single-nucleotide polymorphisms discovery and high-density genetic map construction in cauliflower using specific-locus amplified fragment sequencing. Front Plant Sci 7:334
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31472278), National High Technology Research and Development Program of China (863 Program) (2012AA10A400), and the Shanghai Universities Knowledge Service Platform (ZF1206). We would like to thank the native English-speaking scientists of Elixigen Company.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Niu, D., Du, Y., Wang, Z. et al. Construction of the First High-Density Genetic Linkage Map and Analysis of Quantitative Trait Loci for Growth-Related Traits in Sinonovacula constricta . Mar Biotechnol 19, 488–496 (2017). https://doi.org/10.1007/s10126-017-9768-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-017-9768-2