
Abstract
Coastal systems could be affected by hypoxic events brought about by global change. These areas are essential nursery habitats for several fish species including the common sole (Solea solea L.). Tolerance of fish to hypoxia depends on species and also on their physiological condition and nutritional status. Indeed, high dietary lipid content has been recently shown to negatively impact the resistance of sole to a severe hypoxic challenge. In order to study the molecular mechanisms involved in the early response to hypoxic stress, the present work examined the hepatic transcriptome in common sole fed diets with low and high lipid content, exposed to severe hypoxia. The activity of AMP-activated protein kinase (AMPK) was also investigated through the quantification of threonine-172 phosphorylation in the alpha subunit. The results show that hypoxia consistently regulates several actors involved in energy metabolism pathways and particularly AMPKα, as well as some involved in cell growth and maintenance or unfolded protein response. Our findings reveal that (1) the expression of genes involved in biological processes with high energy cost or implicated in aerobic ATP synthesis was down-regulated by hypoxia, contrary to genes involved in neoglucogenesis or in angiogenesis, (2) the consumption of high lipid induced regulation of metabolic pathways going against this energy saving, and (3) this control was fine-tuned by the regulation of several transcriptomic factors. These results provide insight into the biological processes involved in the hepatic response to hypoxic stress and underline the negative impact of high lipid consumption on the tolerance of common sole to hypoxia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over the past 50 years, the intensification of anthropogenic activities along riversides and coastlines, combined with global warming, has been connected with the increasing duration and severity of environmental hypoxia in many coastal marine ecosystems. Consequently, hypoxia is now considered as one of the most pressing environmental issues worldwide (Wu 2002). Shallow coastal ecosystems are nursery areas for many benthic fish species, and it is believed that hypoxic events can potentially impact them.
A large number of studies have been conducted to examine the physiological consequences and regulatory mechanisms that respond following a reduction in oxygen availability (reviewed by Richards 2009). Although the physiological response to hypoxia varies among fish species (Mandic et al. 2009), it is accepted that they follow a general strategy aimed at inhibiting oxygen- and ATP-demanding metabolic pathways while favoring the activation of oxygen-independent ATP-production pathways (reviewed by Almeida-Val et al. 2006; Bickler and Buck 2007; Lopez-Barneo et al. 2010; Richards 2011). All these studies showed the common feature that this transition requires adequate stores of glucose, generally in the form glycogen, such as that found in the liver (Richards 2011).
The liver plays a central role in synthesizing or converting molecules that are utilized elsewhere to maintain homeostasis and in regulating energy balance. Since the regulations of enzyme activity required for metabolic adaptation to hypoxia have been shown to be related to the differential transcription of mRNA (Semenza et al. 1996), a large-scale investigation of gene expression would improve understanding of the overall impact of hypoxia on fish physiology. Previous studies performed on hepatic tissue have revealed impacts of hypoxia exposure on the expression of genes involved in energy metabolism (i.e., glucose metabolism), cell growth and proliferation, protein degradation, and oxygenase activities (Gracey et al. 2001; Ju et al. 2007; Leveelahti et al. 2011; Everett et al. 2012). As in mammals, hypoxia inducible factors (HIFs) are thought to be involved as transcription factors in the coordination of molecular responses in fish (see review by Nikinmaa and Rees 2005), especially in the liver (Gracey et al. 2001). AMPK, a nutrient and energy sensor that maintains energy homeostasis, is also suggested to play a central role in coordinating the metabolic responses of fish exposed to severe hypoxia (Jibb and Richards 2008). However, numerous key actors and cellular transduction pathways involved in the transcriptional control of metabolism during hypoxia are still unknown.
In the present study, we applied microarray-based gene expression profiling to investigate the short-term transcriptional response of metabolic reprogramming during an acute, severe hypoxia challenge in juvenile common sole (Solea solea). Moreover, phosphorylation of threonine 172 in the alpha subunit, which is a key determinant of AMPK activity (Hardie 2008), was assessed on the same fish. Common sole is a flatfish of the Soleidae family, which is particularly exposed to hypoxia events during juvenile stages because estuaries have been identified as essential nurseries for this species (Le Pape et al. 2003). Common sole can tolerate quite severe drops in ambient oxygenation and does not show a strong avoidance response to hypoxic estuarine environments (Cannas et al. 2007). Previous studies aiming to better characterize the physiological response of common sole to hypoxia revealed a typical metabolic depression (Dalla Via et al. 1994, 1997). Moreover, recent studies have suggested that the dietary lipid content is a factor influencing hypoxia tolerance of common sole (McKenzie et al. 2008; Zambonino-Infante et al. 2013). In particular, Zambonino-Infante et al. (2013) showed that juvenile sole exhibited a lower tolerance to hypoxia when fed a lipid-rich diet. Even though this effect can probably be related to the known low nutritional tolerance of sole species to high lipid ingestion, which induces perturbation of their energy metabolism, the molecular actors and biological processes involved in these regulations are poorly documented. To improve our understanding of these processes, we investigated the hepatic transcriptome in juvenile sole fed diets with low and high lipid contents.
Material and Methods
Feeding Trial, Hypoxia Challenge Stress, and Sampling
Experiments were conducted in strict compliance with the Guide for the Care and Use of Laboratory Animals (National Research Council 2010). One thousand North Sea sole (Solea solea) eggs were purchased from a commercial hatchery (SOLEA BV, Ijmuiden, Netherlands) and brought to the Ifremer larval rearing facility in Brest. Details on larval rearing equipment and conditions can be found in Zambonino-Infante et al. (2013). Eight months post-hatching, 160 individuals were randomly drawn from the rearing tanks, tagged subcutaneously (passive integrated transponder: “PIT-tag”) and distributed among eight 67-L tanks. For two months, these fish were maintained at 16 °C and fed with commercial diets containing either 11 % lipids (BP Finition Label Extrudé, Le Gouessant, France) (l-group) or 20 % lipids (Neo Grower Extra Marin, Le Gouessant, France) (L-group) with four tanks per dietary group.
Prior to the hypoxia challenge test, 74 individuals selected at random (37 from each dietary group) were moved into a single tank (1 m3) and left undisturbed and unfed for 48 h. The blood and liver of 36 of these individuals (18 from each dietary group) were sampled in normoxic conditions for biochemical and molecular analysis, respectively.
The hypoxia challenge was applied to the remaining 38 fishes (19 from each dietary group) and consisted in decreasing water oxygenation from 100 % air saturation to 10 % air saturation within 1 h, followed by a slower descent to 1 % air sat. over the last 30 min. Ambient oxygenation was controlled by bubbling nitrogen at the intake of a submersible pump placed in the tank. After 90 min of hypoxia, fish were removed, identified (PIT-tag reading), and their blood and liver sampled. The resulting four final experimental groups were named as follows: Normoxia l-group (Nl), Normoxia L-group (NL), Hypoxia l-group (Hl), and Hypoxia L-group (HL). The experimental design was therefore a two-way full factorial design between two juvenile dietary lipid contents and two oxygenation conditions. Data related to blood (biochemical) parameters measured in these groups were recently published (Zambonino-Infante et al. 2013).
RNA Extraction and Purification
Liver tissue from 74 individuals was homogenized in 2 ml Extract All Reagent (Eurobio, Courtaboeuf, France). Total RNA was isolated following the manufacturer’s instructions and quantified by measuring absorbance at 260 nm in a spectrophotometer (NanoDrop, Labtech, France). RNA integrity was also evaluated using a Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA) analysis. Following the evaluation of RNA integrity, 71 samples (with RIN > 8) were kept for microarray hybridization: 18 from group Nl, 18 from group NL, 16 from group Hl, and 19 from group HL.
Quantification of Threonine 172 Phosphorylation of AMPKα
Total protein extracts were obtained according to Corporeau and Auffret (2003) from the same 74 liver samples. Briefly, to solubilize proteins, powdered tissues were homogenized in a lysis buffer containing phosphatase and protease inhibitors (150 mM NaCl, 10 mM Tris, pH 7.4, 1 mM EDTA, 1 mM EGTA, 1 % Triton X-100, 0.5 % Igepal, 5 ml phosphatase inhibitor cocktail II, 2 tablets of complete EDTA-free protease inhibitor cocktail; pH 8.8 at 4 °C). Total protein extracts were then quantified using a DC protein assay (Biorad), and protein concentrations were determined quantitatively using 96-well micro-plates (Nunc) and a micro-plate reader connected to KC4 v3 software (Bio-Tek Instruments, Inc).
Prior to quantification of threonine 172 phosphorylation of AMPKα, each protein lysate was adjusted to a final concentration of 3.5 mg/ml by adding lysis buffer. Thr172 phosphorylation of AMPKα was detected by the PathScan® Phospho-AMPKα (Thr172) Sandwich ELISA Kit following manufacturer’s instructions (Cell Signaling Technology, ref #7959). Western blot analysis from protein lysates of sole tissues was previously performed using rat anti-AMPKα (23A3) (rabbit mAb, #2603, Cell Signaling) to ensure that the ELISA assay reported correctly threonine 172 phosphorylation of AMPK in sole. A specific band was detected at approximately 60 kDa, confirming high AMPKα amino-acid sequence identity (>90 %) between sole and rat species (Additional file 1).
Microarray Design
Gene expression profiling of S. solea samples was conducted using an updated version of the Agilent-036353 S. solea DNA microarray (GPL16124). Microarray design was carried out based on 25,252 contigs (isotigs) and 66,026 singletons (with a minimal length of 200 nt) obtained from the Roche 454 sequencing of a normalized cDNA library of sole larval stages (from 1 to 33 days post-hatching) and adult tissues (Ferraresso et al. 2013). All assembled isotigs are stored in the public database Transcriptome Shotgun Assembly Sequence Database (TSA, http://www.ncbi.nlm.nih.gov/genbank/tsa) under accession number GAAQ00000000, while singletons can be directly retrieved from the NCBI Sequence Read Archive (SRA) under accession number SRA058691.
Transcript annotation for both isotigs and singletons was implemented through blastx searches (cut-off e-value of <1.0E-5) against high-quality draft proteomes of Danio rerio, Gasterosteus aculeatus, Oryzias latipes, Takifugu rubripes, Tetraodon nigroviridis, and Homo sapiens, available on the Ensembl Genome Browser (release 56), and against the SWISSPROT database. A schematic representation of the approach used for microarray design is given in Additional file 2. Briefly, all annotated isotigs were screened for redundancy using Ensembl Protein IDs. Two or more transcripts were considered “redundant” when they showed the same annotation as at least three fish species out of five, by considering Ensembl Protein ID of five fish species (D. rerio, G. aculeatus, O. latipes, T. nigroviridis, and T. rubripes), and only the longest was considered for microarray design. The same approach was followed for filtering out redundant singletons. A total of 8,750 isotigs and 5,800 singletons were finally represented in the array.
A total of 14,701 probes, targeting 14,531 transcripts, were successfully designed; 8,918 of these had been previously employed on the first version of the array (GEO acc: GPL16124). Probe sequences and further details on the microarray platform can be found in the GEO repository (http://www.ncbi.nlm.nih.gov/geo/) under accession number GPL16714.
Microarray Hybridization and Data Analysis
Sample labeling and hybridization were performed according to the Agilent One-Color Microarray-Based Gene Expression Analysis protocol for 8 × 15 K slide format.
Briefly, for each sample, 100 ng of total RNA was linearly amplified and labeled with Cy3-dCTP. A mixture of 10 different viral poly-adenylated RNAs (Agilent Spike-In Mix) was added to each RNA sample before amplification and labeling, to monitor microarray analysis workflow. Labeled cRNA was purified with a Qiagen RNeasy Mini Kit, and sample concentration and specific activity (pmol Cy3/μg cRNA) were measured in a NanoDrop® ND-1000 spectrophotometer. A total of 600 ng labeled cRNA was prepared for fragmentation by adding 5 μl 10× blocking agent and 1 μl 25× fragmentation buffer, heated to 60 °C for 30 min, and finally diluted by addition of 25 μl 2× GE hybridization buffer. A volume of 40 μl hybridization solution was then dispensed in the gasket slide and assembled with the microarray slide (each slide containing eight arrays). Slides were incubated for 17 h at 65 °C in an Agilent hybridization oven, then removed from the hybridization chamber, quickly submerged in GE Wash Buffer 1 for disassembly of the slides, and then washed in GE wash buffer 1 for approximately 1 min, followed by one additional wash in pre-warmed (37 °C) GE wash buffer 2.
Hybridized slides were scanned at 5 μm resolution using an Agilent G2565BA DNA microarray scanner. Default settings were modified to scan each slide twice at two different sensitivity levels (XDR Hi 100 % and XDR Lo 10 %). The two linked images generated were analyzed together, and the data were extracted and background subtracted using the standard procedures in the Agilent Feature Extraction (FE) Software version 9.5.1. Cyclic loess normalization was performed using R statistical software. Spike-in control intensities were used to monitor the normalization procedure. Raw and normalized fluorescence data of all microarray experiments were deposited in the GEO database under accession numbers GSE44579.
Statistical Analysis
Two-way ANOVA (stat soft) was used to compare the phosphorylation levels of AMPKα between groups. The microarray data were also analyzed by two-way ANOVA using Tmev (TIGR MultiExperiment Viewer) statistical software, and gene expression was considered significantly different when p-value < 0.005. No multiple test correction (i.e., Bonferroni) was employed as previous analyses indicated that such corrections could be over-conservative (Leaver et al. 2008). However, in order to minimize the type 1 error and related false discovery rates (FDR), p-value was fixed at 0.005. Significant enrichment of GO biological process categories was tested using EASE software (version 2.0). Benjamini correction was applied for statistical analysis related to GO enrichment.
Results and Discussion
Impact of Hypoxic Challenge
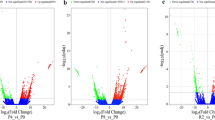
The main objective of the present study was to investigate the short-term response of the liver transcriptome to an acute hypoxic challenge in common sole fed diets of different lipid content. The reliability of datasets was confirmed by the reproducibility of the signal obtained from redundant transcript (data not shown). By examining genes regulated by the “hypoxia” factor, we revealed the molecular actors and related processes underlying the physiological response of common sole to hypoxia in liver tissue. ANOVA revealed that 1,202 genes (FDR < 7 %, with a p-value of 0.005) were significantly regulated following hypoxia, amounting to 8 % of the total spotted genes (Table 1 and Fig. 1). Among these regulated genes, 514 were up-expressed under hypoxic conditions, and 688 were down-expressed (Additional file 3). Gene ontology enrichment analysis, which enables the identification of GO terms significantly enriched in the input entity list when compared to the whole array dataset, was performed providing evidence for which biological processes may be particularly altered by hypoxia factor. Four significant GO terms, all interrelated, were identified: carbohydrate metabolism, glucose metabolism, energy pathways, and molecular process related to oxidoreductase activity (Table 2). Based on the GO categorization, part of the up-regulated genes was related to the gluconeogenesis pathway, regulation of cell growth and maintenance, sulfur amino acid biosynthesis, blood vessel development, or transcription regulator activity; among down-regulated genes, these were mitochondrial ATP synthesis, tricarboxylic acid cycle (TCA) intermediate metabolism, glycolysis, lipid biosynthesis, and protein folding (Table 3). The present results revealed an important gene-based metabolic reprogramming characterized by the down-regulation of energy-demand and aerobic energy-supply pathways associated with an induction of processes involved in the supply of anaerobic metabolic substrates and cell rescue.

Venn diagram showing the number of genes significantly up- and down-regulated by hypoxia and lipid factors
Inhibition of Genes Related to Aerobic ATP Supply and Energy Consumption
We found numerous genes involved in energy metabolism down-regulated by hypoxia (Table 3). These include four genes involved in ATP synthesis-coupled electron transport (NADH dehydrogenase ubiquinones: five NDUF genes) and four genes involved in the TCA cycle (ACLY, GAD1, IDH1, and ME1). These down-regulations are consistent with the known reduction of energy production by aerobic metabolic pathways in fish subjected to hypoxic conditions (Krumschnabel et al. 2000). Such down-regulations are also well documented in fish (Wright et al. 1989; Martinez et al. 2006), including common sole (Dalla Via et al. 1994; Van den Thillart et al. 1994), and inhibition of aerobic pathways has been seen to be concomitant with an activation of anaerobic ATP-generating pathways which use glucose as the main substrate. In agreement with these previous results, we found that several genes up-regulated by hypoxia are involved in the key steps of the gluconeogenesis pathway (PCK1 and G6PC) and in the metabolism of glucogenic amino acid, such as the transaminases (AADAT, GOT1, OAT, HAL, and TAT) (Table 3). Our transcriptomic data, which suggest the stimulation of the expression of genes implicated in glucose synthesis through gluconeogenesis, are consistent with the significant hypoxia-induced increase of glycemia that we measured in these fish (Zambonino-Infante et al. 2013). They are also in agreement with previous transcriptomic results on the liver of longjaw mudsucker (Gracey et al. 2001). However, contrary to data obtained by Gracey and collaborators revealing a stimulation of genes involved in hepatic glycolysis, we observed a significant down-regulation of several genes (ENO1, G6PD, HK2, LDHA, LDHB, PFKL, PGD, PGK1, PKM2, and TALDO1) involved in glucose catabolism pathways, including glycolysis (Table 3). The inhibition of glucose catabolism in the liver points to a strategy of relocalization of energy supply in sole. This strategy could consist in maintaining high blood glucose levels for anaerobic ATP production in priority organs, such as the brain and heart, when survival is at stake.
Interestingly, we also found under hypoxic conditions higher hepatic levels of threonine 172 phosphorylation of AMP-activated protein kinase (AMPKα), which is an energy sensor protein kinase that plays a key role in maintaining cellular energy balance (Fig. 2). Considering that this phosphorylation is a key determinant of AMPK activity (Hardie 2008), our data are consistent with the stimulation of AMPK activity observed by Jibb and Richards (2008) in severely hypoxic goldfish. It is well documented that AMPK inhibits energetically expensive anabolic processes, such as protein, glycogen, or fatty acid synthesis and cell growth and proliferation, in response to reduction of intracellular ATP levels (Mihaylova and Shaw 2011). In agreement with the present AMPKα stimulation, we observed 19 genes involved in lipid biosynthesis down-regulated and several genes implicated in the suppression of cell growth and proliferation, such as IGFBP-1, GRB10, DUSP1 and DUSP6, TOB1, and BTG-1 (Table 3), up-regulated under hypoxic conditions. While inductions of IGFBP-1 and GRB10 have been shown to inhibit in vivo IGF action, particularly under hypoxic conditions (Tazuke et al. 1998; Liu and Roth 1995; Kajimura et al. 2005), mitogen-activated proteins DUSP1 and DUSP 6 are known to inactivate the ERK group of MAP kinase involved in cell growth stimulation. The stimulation of IGFBP-1 expression that limits IGF-dependent decrease of growth hormone receptor (GHR) expression (Min et al. 1996) can be related to the increase in hepatic GHR mRNA levels that we observed during hypoxia (Table 3). TOB1 and BGT-1 are also known to suppress growth through their anti-proliferative function (Ho et al. 2010; Kamaid and Giraldez 2008). The impact of hypoxia on the expression of genes involved in cell growth and proliferation supports the view that fish experiencing reduced oxygen availability must reallocate energy from growth toward life-sustaining processes.

Quantification of threonine 172 phosphorylation in the alpha subunit of AMPK in the Normoxia l-group (Nl), Normoxia L-group (NL), Hypoxia l-group (Hl), and Hypoxia L-group (HL). Different letters (a, b) indicate a significant difference between the groups (p < 0.05)
Induction of Vascularization
Among the regulated genes, we found some actors involved in the regulation of vascular endothelial cells (Table 3). GRB10, known to negatively regulate the insulin pathway, is also involved in angiogenesis by regulating the KDR/VEGFR-2 signaling pathway. The up-regulation of this gene suggests a stimulation of vascular endothelial cell development. In line with this view, we also noted the stimulation of several actors involved in blood vessel development including ERBB4 and EGFR genes, both of which play an essential role as a cell surface receptor of EGF (Russell et al. 1999). ERBB have been shown to play a key role in the regulation of angiogenesis by inducing the vascular endothelial growth factor (VEGF) (Yen et al. 2002). Even though we did not observe any significant differential expression of VEGF in the present work, we found a positive regulation by hypoxia challenge of PRKCA, PDGFRB, FOXF1, JAG1, RAMP2, and STAB1 (Table 3), all known to be involved in blood vessel development (Adachi and Tsujimoto 2002; Ichikawa-Shindo et al. 2008; Stankiewicz et al. 2009; Wang et al. 2002; Zimrin et al. 1996). This stimulation of blood vessel formation during hypoxia, which has been widely documented in vertebrates including fish species (reviewed by Nikinmaa and Rees 2005), reflects the need to optimize oxygen supply to tissues. However, contrary to what has been shown in previous studies performed in rainbow trout by Marinsky et al. (1990) or in the euryoxic fish Gillichthys mirabilis by Gracey et al. (2001), we could not find any significant regulation of actors involved in iron or oxygen binding. It is likely that these regulations, even if they exist in the liver of common sole, do not represent the first line of adaptive strategy at the transcriptomic level.
As they highlight the impact of a hypoxic stress on several processes involved in the regulation of cellular metabolism and growth, as well as oxygen delivery to tissue cells, these data are consistent with previous reports performed on the liver of different fish species (Gillichthys mirabilis, Gracey et al. 2001; Oryzias latipes, Ju et al. 2007; Gasterosteus aculeatus, Leveelahti et al. 2011; Fundulus grandis, Everett et al. 2012).
Regulation of Redox Potential
It is noteworthy that some of the hypoxia-stimulated genes are also involved in sulfur amino acid biosynthesis (CBS and CTH) and in the glutathione metabolism process (GGT1) (Table 3). This regulation probably allows an optimal intracellular glutathione level to be maintained, contributing to cellular antioxidant defense mechanism under hypoxic conditions. In agreement with the above, our transcriptomic data also revealed the differential expression of 56 genes with oxidoreductase activity (Table 2), reinforcing the link between reduced oxygen availability and the regulation of redox potential. Altogether, such transcriptomic features can be related to the increase in mitochondrial reactive oxygen species (ROS) production classically observed in hypoxia (Chandel et al. 1998; Mansfield et al. 2005).
Large-Scale Regulation of the Hepatic Transcriptome by Transcriptomic Factors
As mentioned in Table 1, our results indicate that a large number of genes (n = 1,202) are significantly regulated 90 min after the onset of hypoxia. The immediate large-scale effect of hypoxia on the hepatic transcriptome may be partly explained by early regulation of the expression of several transcription factors (i.e., JUN, FOS, EGR1, HIF3, CREB, ATF7IP, and CEBPD) listed in Table 3. These transcription factors are indeed involved in a cascade of regulation involving a battery of genes that act in concert to facilitate the supply of oxygen and nutrients, regulate energy metabolism, and promote cell survival and growth control (Hochachka et al. 1996; Cummins and Taylor 2005). This result demonstrates that, in addition to the regulations occurring at the post-transcriptomic level (van den Beucken et al. 2011), a severe hypoxic stress has a rapid and profound effect on the hepatic transcriptome.
Activation of the Unfolded Protein Response (UPR)
We found several genes involved in protein folding (9 genes) or related to endoplasmic reticulum (RE) (21 genes) down-regulated by hypoxia challenge (Table 3). In vertebrates, it is well documented that regulation of the protein folding process is associated with RE stress trigger UPR (Kim et al. 2008). Interestingly, HSPA5 was shown to be up-regulated following hypoxia (Table 3). HSPA5 is a key calcium-dependent chaperone involved in the setting off of the downstream signaling of the UPR through its association with IRE1, ATF6, and PERK (Kim et al. 2008). In a similar way as in the results obtained by Tagliavacca et al. (2012) and Kim et al. (2008) in mammals, the present data suggest that these pathways were also triggered by hypoxia in fish. In mammals, it is well documented that the PERK pathway is initiated by the phosphorylation of eIF2α, thereby blocking protein translation and activating the transcription factor ATF4. ATF4 controls the expression of genes involved in redox balance, amino acid metabolism, protein folding, and apoptosis (Ameri and Harris 2008). In the present work, we observed up-regulation of challenge target genes of ATF4 such as the previously mentioned folding protein HSPA5 and the amino acid transporter SLC7A3 as well as IGFBP-1 and GRB10, which are implicated in cell growth mechanisms (Luo et al. 2003; Harding et al. 2003) (Table 3). This suggests that the PERK pathway is also activated during hypoxia in sole. With respect to the other UPR pathways, it is known that ATF6 and XBP1 in mammals are able to transactivate genes encoding chaperones and proteins involved in endoplasmic reticulum-associated degradation (ERAD) (Jaeger et al. 2012; Kim et al. 2008). The stimulation of the ATF6-target gene Herpud1 (Table 3), involved in ERAD (Ma and Hendershot 2004), also suggests the activation of the ATF6 pathway within the 90 min of hypoxia tested in the present study.
While UPR consists of mechanisms decreasing the synthesis of proteins and their influx into the ER, we found only a few genes involved in this process that were regulated during environmental hypoxia. This observation suggests that the main mechanisms responsible for the decrease in protein synthesis shortly after the initiation of hypoxia require post-transcriptional regulation.
Our data revealed that most of the molecular actors involved in ER stress, as they were described for mammals, can also be found in common sole. Moreover, these data are totally consistent with a recent study of Ishikawa et al. (2011) indicating that the process of UPR is conserved across vertebrates including fish.
Impact of Dietary Lipid Content
The second objective of this work was to determine the biological processes underlying the lower tolerance of sole to hypoxia when fish were fed a lipid-rich diet (Zambonino-Infante et al. 2013). Statistical analysis revealed 801 genes (FDR < 10 %, with a p-value of 0.005) regulated by the dietary lipid content, whatever the oxygenation conditions (Table 1; Fig. 1). These included 424 genes down-expressed and 377 genes up-expressed in fish fed the high-lipid diet (Additional file 3). Interestingly, we found only 200 genes with expression differentially affected by hypoxia depending on the dietary lipid content. It is noteworthy that gene ontologies related to macromolecule biosynthesis and protein folding, mitochondrial ATP synthesis, and defense activity were significantly enriched within genes regulated by diet condition whatever the oxygenation conditions (Table 4).
Regulation of Glucose Metabolism
We found that hepatic nuclear factor 6 (ONECUT1) was stimulated in fish fed the high-fat diet (Table 5). It is known that ONECUT1 is involved in the regulation of the transcription of gluconeogenic enzymes such as glucose-6-phosphatase (G6PC) (Streeper et al. 2001). In line with this, we also observed a stimulation of TCA enzymes involved in gluconeogenesis (MDH1 and MDH2) in the same dietary group (Table 5). Together with all the mechanisms involved in high-fat-diet-induced diabetes, this result could explain the higher glycemia rate observed in fish fed the high-lipid diet (Zambonino-Infante et al. 2013).
Stimulation of an ATP-Expensive Anabolic Process and Aerobic ATP Synthesis
Analysis of GO revealed that up-regulated genes were also related to protein biosynthesis and folding (61 genes, see Table 5). The stimulation of genes involved in protein synthesis and maturation could be due to an increased need for lipoproteins, required to transport the overload of dietary lipids (Lim et al. 2009; Yildirim-Aksoy et al. 2009). This result contrasts with data obtained in mice fed a high-fat diet, where a down-regulation of protein synthesis was observed (Deldicque et al. 2010; Oyadomari et al. 2008). This discrepancy may be likened to the low lipid tolerance of flatfish species (Borges et al. 2009). As mentioned by Zambonino-Infante et al. (2013), the relative difficulty that sole have at handling high dietary lipid contents may have a metabolic cost. In agreement with this idea, the present study revealed some up-regulated genes involved in oxidative phosphorylation and ATP biosynthesis (NDUFs and ATP synthases, Table 5). Globally, around 10 % of the genes up- and down-regulated by hypoxia factor were found to be down- and up-regulated by lipid factor, respectively (Fig. 1); GO over-represented within those genes being related to mitochondria and hydrogen ion transporter activity (Table 6). These data suggest an increased activity of aerobic ATP production in response to high-fat feeding in common sole, probably through the positive regulation of fatty acid beta-oxidation and the TCA pathway.
The stimulation of the ATP-expensive anabolic process, supported by the lower rates, although not significant (p = 0.07), of AMPKα phosphorylation (Fig. 2), goes against the principle that hypoxic fish should save oxygen. These regulations were unrelated to the oxygen conditions and could therefore explain the impaired tolerance to hypoxia in fish fed the high-lipid diet.
Low Significant Hypoxia × Lipid Interaction on the Hepatic Transcriptome
Our statistical analysis revealed only a slight interaction between hypoxia and lipid factors on the hepatic transcriptome (i.e., 200 genes, see Table 1 and Additional file 3). However, biological data mining resulting from this analysis is ambiguous since FDR is expected to be around 30 % using a p-value of 0.005. Moreover, no gene ontology was enriched among the 200 genes.
Impact of Dietary Lipid Content on Defense Activities
Finally, our transcriptomic data revealed that feeding fish with a high-lipid-content diet down-regulated genes associated with blood coagulation, immune response, and homeostasis (Table 5). These data confirm earlier studies that demonstrated an effect of the dietary lipid content on the fish immune system, particularly on complement activity (Geay et al. 2011). Together with the impact of high lipid ingestion on the energy metabolism of common sole, this last result suggests deleterious effects on the processes of defense and further undermines the tolerance of this fish species to lipids.
Conclusion
The present data reveal new relevant information about the short-term effect of acute hypoxia on the fish liver transcriptome. In particular, the data highlight a finely tuned regulation of different biological processes, including metabolic pathways and UPR response, resulting in metabolic depression. Our results revealed that the regulation of these processes could be related to the stimulation of several transcription factors, explaining the large-scale regulation of the hepatic transcriptome. Moreover, our data provide valuable insight on the influence of high lipid consumption on liver metabolic pathways. While several other tissues (i.e., muscle, heart, and brain), as well as post-transcriptomic levels of regulations, are certainly affected by lipid content in diet, we can hypothesize that the regulations induced by high-lipid diets in liver are detrimental to the energy saving required for cellular homeostasis under hypoxia. This effect could partly contribute to explaining why fish fed high level of fat exhibited lower tolerance to hypoxia compared to those fed a low-lipid diet. It is indeed accepted that hypoxia tolerance is largely based on an organism’s ability to down-regulate ATP production and consumption in a coordinated way during energy-limiting conditions (Krumschnabel et al. 2000). It is suggested that climate warming will induce changes on zooplankton communities and especially lipid-storing species, which would affect the lipid flux of the entire system (Lee et al. 2006). Assessing the molecular mechanisms underlying the effects of dietary lipid content on hypoxia tolerance in common sole is crucial for a better understanding of the effects that global change will have on the physiology of this specific taxon, on its life traits, and on the possible consequences in terms of population.
References
Adachi H, Tsujimoto M (2002) FEEL-1, a novel scavenger receptor with in vitro bacteria-binding and angiogenesis-modulating activities. J Biol Chem 277:34264–34270
Almeida-Val VMF, Chipari-Gomes AR, Lopes NP (2006) Metabolic and physiological adjustments to low oxygen and high temperature in fishes of the Amazon. In: Adalberto Luis V, Almeida-Val VMF, Randall DJ (eds) The physiology of tropical fishes. Elsevier, London
Ameri K, Harris AL (2008) Activating transcription factor 4. Int J Biochem Cell Biol 40:14–21
Bickler, P.E. & Buck, L.T. (2007). Hypoxia tolerance in reptiles, amphibians, and fishes: life with variable oxygen availability. Ann Rev Physiol 69:145–170
Borges P, Oliveira B, Casal S, Dias J, Conceicao L, Valente LMP (2009) Dietary lipid level affects growth performance and nutrient utilisation of Senegalese sole (Solea senegalensis) juveniles. Br J Nutr 102:1007–1014
Cannas M, Bayle A, Wing OLC, Domenici P, Lefrancois C (2007) Behaviour and physiology of the startle response in common sole (Solea solea) exposed to hypoxia. Comp Biochem Physiol A Mol Integr Physiol 146:S85
Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT (1998) Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A 95:11715–11720
Corporeau C, Auffret M (2003) In situ hybridisation for flow cytometry: a molecular method for monitoring stress-gene expression in hemolymph cells of oysters. Aquat Toxicol 64:427–435
Cummins EP, Taylor CT (2005) Hypoxia-responsive transcription factors. Pflugers Arch - Eur J Physiol 450:363–371
Dalla Via J, Vandenthillart G, Cattani O, Dezwaan A (1994) Influence of long-term hypoxia exposure on the energy-metabolism of Solea solea. 2. Intermediary metabolism in blood, liver and muscle. Mar Ecol Prog Ser 111:17–27
Dalla Via J, Vandenthillart G, Cattani O, Cortesi P (1997) Environmental versus functional hypoxia/anoxia in sole Solea solea: the lactate paradox revisited. Mar Ecol Prog Ser 154:79–90
Deldicque L, Cani PD, Philp A, Raymackers J-M, Meakin PJ, Ashford MLJ, Delzenne NM, Francaux M, Baar K (2010) The unfolded protein response is activated in skeletal muscle by high-fat feeding: potential role in the downregulation of protein synthesis. Am J Physiol Endocrinol Metab 299:E695–E705
Everett MV, Antal CE, Crawford DL (2012) The effect of short-term hypoxic exposure on metabolic gene expression. J Exp Zool A Ecol Genet Physiol 317A:9–23
Ferraresso S, Bonaldo A, Parma L, Cinotti S, Massi P, Bargelloni L, Gatta PP (2013) Exploring the larval transcriptome of the common sole (Solea solea L.). BMC Genomics 14:315–337
Geay, F., Ferraresso, S., Zambonino-Infante, J.L., Bargelloni, L., Quentel, C., Vandeputte, M., Kaushik, S., Cahu, C.L. & Mazurais, D. (2011). Effects of the total replacement of fish-based diet with plant-based diet on the hepatic transcriptome of two European sea bass (Dicentrarchus labrax) half-sibfamilies showing different growth rates with the plant-based diet. BMC Gen 12:522–540
Gracey AY, Troll JV, Somero GN (2001) Hypoxia-induced gene expression profiling in the euryoxic fish Gillichthys mirabilis. Proc Natl Acad Sci U S A 98:1993–1998
Hardie DG (2008) AMPK: a key regulator of energy balance in the single cell and the whole organism. Int J Obes (Lond) 32(Suppl 4):S7–S12
Harding HP, Zhang YH, Zeng HQ, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11:619–633
Ho KJ, Do NL, Otu HH, Dib MJ, Ren X, Enjyoji K, Robson SC, Terwilliger EF, Karp SJ (2010) Tob1 is a constitutively expressed repressor of liver regeneration. J Exp Med 207:1197–1208
Hochachka PW, Buck LT, Doll CJ, Land SC (1996) Unifying theory of hypoxia tolerance: molecular metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci U S A 93:9493–9498
Ichikawa-Shindo Y, Sakurai T, Kamiyoshi A, Kawate H, Linurna N, Yoshizawa T, Koyama T, Fukuchi J, Limuro S, Moriyama N, Kawakami H, Murata T, Kangawa K, Nagai R, Shindo T (2008) The GPCR modulator protein RAMP2 is essential for angiogenesis and vascular integrity. J Clin Investig 118:29–39
Ishikawa T, Taniguchi Y, Okada Z, Takeda S, Mori K (2011) Vertebrate unfolded protein response: mammalian signaling pathways are conserved in medaka fish. Cell Struc Func 36:247–259
Jaeger R, Bertrand MJM, Gorman AM, Vandenabeele P, Samali A (2012) The unfolded protein response at the crossroads of cellular life and death during endoplasmic reticulum stress. Biol Cell 104:259–270
Jibb LA, Richards JG (2008) AMP-activated protein kinase activity during metabolic rate depression in the hypoxic goldfish, Carassius auratus. J Exp Biol 211:3111–3122
Ju Z, Wells MC, Heater SJ, Walter RB (2007) Multiple tissue gene expression analyses in Japanese medaka (Oryzias latipes) exposed to hypoxia. Comp Biochem Physiol C Toxicol Pharmacol 145:134–144
Kajimura S, Aida K, Duan CM (2005) Insulin-like growth factor-binding protein-1 (IGFBP-1) mediates hypoxia-induced embryonic growth and developmental retardation. Proc Natl Acad Sci U S A 102:1240–1245
Kamaid A, Giraldez F (2008) Btg1 and Btg2 gene expression during early chick development. Dev Dyn 237:2158–2169
Kim I, Xu W, Reed JC (2008) Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 7:1013–1030
Krumschnabel G, Schwarzbaum PJ, Lisch J, Biasi C, Wieser W (2000) Oxygen-dependent energetics of anoxia-tolerant and anoxia-intolerant hepatocytes. J Exp Biol 203:951–959
Le Pape O, Chauvet F, Mahevas S, Lazure P, Guerault D, Desaunay Y (2003) Quantitative description of habitat suitability for the juvenile common sole (Solea solea, L.) in the Bay of Biscay (France) and the contribution of different habitats to the adult population. J Sea Res 50:139–149
Leaver MJ, Villeneuve LA, Obach A, Jensen L, Bron JE, Tocher DR, Taggart JB (2008) Functional genomics reveals increases in cholesterol biosynthetic genes and highly unsaturated fatty acid biosynthesis after dietary substitution of fish oil with vegetable oils in Atlantic salmon (Salmo salar). BMC Genomics 9:299–315
Lee RF, Hagen W, Kattner G (2006) Lipid storage in marine zooplankton. Mar Ecol Prog Ser 307:273–306
Leveelahti L, Leskinen P, Leder EH, Waser W, Nikinmaa M (2011) Responses of threespine stickleback (Gasterosteus aculeatus, L) transcriptome to hypoxia. Comp Biochem Physiol D Gen Proteome 6:370–381
Lim C, Yildirim-Aksoy M, Li MH, Welker TL, Klesius PH (2009) Influence of dietary levels of lipid and vitamin E on growth and resistance of Nile tilapia to Streptococcus iniae challenge. Aquaculture 298:76–82
Liu F, Roth RA (1995) GRB-IR—a SH2-domain-containing protein that binds to the insulin-receptor and inhibits its function. Proc Natl Acad Sci U S A 92:10287–10291
Lopez-Barneo J, Nurse CA, Nilsson GE, Buck LT, Gassmann M, Bogdanova AY (2010) First aid kit for hypoxic survival: sensors and strategies. Physiol Biochem Zool 83:753–763
Luo SZ, Baumeister P, Yang SJ, Abcouwer SF, Lee AS (2003) Induction of Grp78/BiP by translational block—activation of the Grp78 promoter by ATF4 through an upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J Biol Chem 278:37375–37385
Ma YJ, Hendershot LM (2004) Herp is dually regulated by both the endoplasmic reticulum stress-specific branch of the unfolded protein response and a branch that is shared with other cellular stress pathways. J Biol Chem 279:13792–13799
Mandic M, Todgham AE, Richards JG (2009) Mechanisms and evolution of hypoxia tolerance in fish. Proc R Soc B Biol Sci 276:735–744
Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, Simon MC (2005) Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab 1:393–399
Marinsky CA, Houston AH, Murad A (1990) Effect of hypoxia on hemoglobin isomorph abundances in rainbow-trout, Salmo gairdneri. Can J Zool-Revue Canadienne De Zoologie 68:884–888
Martinez ML, Landry C, Boehm R, Manning S, Cheek AO, Rees BB (2006) Effects of long-term hypoxia on enzymes of carbohydrate metabolism in the Gulf killifish, Fundulus grandis. J Exp Biol 209:3851–3861
Mckenzie DJ, Lund I, Pedersen PB (2008) Essential fatty acids influence metabolic rate and tolerance of hypoxia in Dover sole (Solea solea) larvae and juveniles. Mar Biol 154:1041–1051
Mihaylova MM, Shaw RJ (2011) The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 13:1016–1023
Min SH, Mackenzie DDS, Breier BH, Mccutcheon SN, Gluckman PD (1996) Responses of young energy-restricted sheep to chronically administered insulin-like growth factor I (IGF-I): evidence that IGF-I suppresses the hepatic growth hormone receptor. Endocrinology 137:1129–1137
National-Research-Council. Guide for the care and use of the laboratory animals. Washington D.C.:National Academic Press, 2010.
Nikinmaa M, Rees BB (2005) Oxygen-dependent gene expression in fishes. Am J Physiol Regul Integr Comp Physiol 288:R1079–R1090
Oyadomari S, Harding HP, Zhang Y, Oyadomari M, Ron D (2008) Dephosphorylation of translation initiation factor 2 alpha enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab 7:520–532
Richards JG (2009) Metabolic and molecular responses of fish to hypoxia. In: Richards JG, Farrell AP, Brauner CJ (eds) Hypoxia. Elsevier, San Diego
Richards JG (2011) Physiological, behavioral and biochemical adaptations of intertidal fishes to hypoxia. J Exp Biol 214:191–199
Russell KS, Stern DF, Polverini PJ, Bender JR (1999) Neuregulin activation of ErbB receptors in vascular endothelium leads to angiogenesis. Am J Physiol Heart Circ Physiol 277:H2205–H2211
Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A (1996) Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem 271:32529–32537
Stankiewicz P, Sen P, Bhatt SS, Storer M, Xia Z, Bejjani BA, Ou Z, Wiszniewska J, Driscoll DJ, Bolivar J, Bauer M, Zackai EH, Mcdonald-Mcginn D, Nowaczyk MMJ, Murray M, Shaikh TH, Martin V, Tyreman M, Simonic I, Willatt L, Paterson J, Mehta S, Rajan D, Fitzgerald T, Gribble S, Prigmore E, Patel A, Shaffer LG, Carter NP, Cheung SW, Langston C, Shaw-Smith C (2009) Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet 84:780–791
Streeper RS, Hornbuckle LA, Svitek CA, Goldman JK, Oeser JK, O’brien RM (2001) Protein kinase A phosphorylates hepatocyte nuclear factor-6 and stimulates glucose-6-phosphatase catalytic subunit gene transcription. J Biol Chem 276:19111–19118
Tagliavacca L, Caretti A, Bianciardi P, Samaja M (2012) In vivo up-regulation of the unfolded protein response after hypoxia. Biochimica Et Biophysica Acta-General Subjects 1820:900–906
Tazuke SI, Mazure NM, Sugawara J, Carland G, Faessen GH, Suen LF, Irwin JC, Powell DR, Giaccia AJ, Giudice LC (1998) Hypoxia stimulates insulin-like growth factor binding protein 1 (IGFBP-1) gene expression in HepG2 cells: a possible model for IGFBP-1 expression in fetal hypoxia. Proc Natl Acad Sci U S A 95:10188–10193
Van Den Beucken T, Magagnin MG, Jutten B, Seigneuric R, Lambin P, Koritzinsky M, Wouters BG (2011) Translational control is a major contributor to hypoxia induced gene expression. Radiother Oncol 99:379–384
Van Den Thillart G, Dalla Via J, Vitali G, Cortesi P (1994) Influence of long-term hypoxia exposure on the energy-metabolism of Solea solea.1. Critical O2 levels for aerobic and anaerobic metabolism. Mar Ecol Prog Ser 104:109–117
Wang AH, Nomura M, Patan S, Ware JA (2002) Inhibition of protein kinase C alpha prevents endothelial cell migration and vascular tube formation in vitro and myocardial neovascularization in vivo. Circ Res 90:609–616
Wright PA, Perry SF, Moon TW (1989) Regulation of hepatic gluconeogenesis and glycogenolysis by cathecholamines in rainbow-trout during environmental hypoxia. J Exp Biol 147:169–188
Wu RSS (2002) Hypoxia: from molecular responses to ecosystem responses. Mar Pollut Bull 45:35–45
Yen L, Benlimame N, Nie ZR, Xiao DZ, Wang TQ, Al Moustafa AE, Esumi H, Milanini J, Hynes NE, Pages G, Alaoui-Jamali MA (2002) Differential regulation of tumor angiogenesis by distinct ErbB homo- and heterodimers. Mol Biol Cell 13:4029–4044
Yildirim-Aksoy M, Lim C, Shelby R, Klesius PH (2009) Increasing fish oil levels in commercial diets influences hematological and immunological responses of channel catfish, Ictalurus punctatus. J World Aquacult Soc 40:76–86
Zambonino-Infante J, Claireaux G, Ernande B, Jolivet A, Quazuguel P, Sévère A, Huelvan C, Mazurais D (2013) Hypoxia tolerance of common sole juveniles depends on dietary regime and temperature at the larval stage: evidence for environmental conditioning. Proc R Soc B. doi:10.1098/rspb.2012.3022
Zimrin AB, Pepper MS, Mcmahon GA, Nguyen F, Montesano R, Maciag T (1996) An antisense oligonucleotide to the notch ligand jagged enhances fibroblast growth factor-induced angiogenesis in vitro. J Biol Chem 271:32499–32502
Acknowledgments
We would like to thank P. Quazuguel for his excellent assistance in maintaining the fish and C. Huelvan and M.M. Le Gall for their assistance with sampling. We are also grateful to C. Quere for their assistance with AMPK analysis and to H. McCombie for English language editing. This work was supported by the SINGER-POLIGNAC foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
David Mazurais and Serena Ferraresso contributed equally to this paper.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Additional file 1
Western blot analysis from sole tissues performed using Rat Anti-AMPKα (JPEG 67 kb)
Additional file 2
Schematic representation of the approach used for microarray design (JPEG 42 kb)
Additional file 3
Full list of genes regulated by “hypoxia”, “lipid” and “hypoxia” × “lipid” factors interaction. (TXT 18 kb)
Rights and permissions
About this article
Cite this article
Mazurais, D., Ferraresso, S., Gatta, P.P. et al. Identification of Hypoxia-Regulated Genes in the Liver of Common Sole (Solea solea) Fed Different Dietary Lipid Contents. Mar Biotechnol 16, 277–288 (2014). https://doi.org/10.1007/s10126-013-9545-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-013-9545-9