
Abstract
Marine scallops, with extended planktonic larval stages which can potentially disperse over large distances when advected by marine currents, are expected to possess low geographical differentiation. However, the sessile lifestyle as adult tends to form discrete “sea beds” with unique population dynamics and structure. The narrow distribution of Zhikong scallop (Chlamys farreri), its long planktonic larval stage, and the extremely hydrographic complexity in its distribution range provide an interesting case to elucidate the impact of marine currents on geographical differentiation for marine bivalves at a fine geographical scale. In this study, we analyzed genetic variation at nine microsatellite DNA loci in six locations throughout the distribution of Zhikong scallop in the Northern China. Very high genetic diversity was present in all six populations. Two populations sampled from the same marine gyre had no detectable genetic differentiation (F ST = 0.0013); however, the remaining four populations collected from different marine gyres or separated by strong marine currents showed low but significant genetic differentiation (F ST range 0.0184–0.0602). Genetic differentiation was further analyzed using the Monmonier algorithm to identify genetic barriers and using the assignment test conducted by software GeneClass2 to ascertain population membership of individuals. The genetic barriers fitting the orientation of marine gyres/currents were clearly identified, and the individual assignment analysis indicated that 95.6% of specimens were correctly allocated to one of the six populations sampled. The results support the hypothesis that significant population structure is present in Zhikong scallop at a fine geographical scale, and marine currents can be responsible for the genetic differentiation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In contrast to terrestrial species, marine organisms are usually expected to show very low geographical differentiation. This mainly attributes to the lack of physical barriers to genetic exchange in “open” oceans and their biological characteristics such as large population size, wide range of distribution, prolific fecundity, and long planktonic larval phases (Palumbi 1992; Nielsen and Kenchington 2001; Luttikhuizen et al. 2003; Baus et al. 2005; Kenchington et al. 2006). However, due to the complexity of marine environments and the diverse life history stages of marine organisms, more complicated factors including historical vicariance events, marine currents and gyres, bays and islands, water temperature and salinity, habitat discontinuity, mobility of marine species in different developmental phases, and even anthropogenic activities can affect the geographical differentiation of different local populations and thereby form unique population dynamics (Palumbi 1992; Edmands and Potts 1997; Collin 2001; Nielsen and Kenchington 2001; Luttikhuizen et al. 2003; Baus et al. 2005; Kenchington et al. 2006; Rose et al. 2006). Based on the cases studied to date, most species in the marine realm share two common properties that are responsible for genetic differentiation among different geographical populations: the dispersal abilities of marine organisms (e.g., Bohonak 1999; Riginos and Nachman 2001) and advection by marine currents (e.g. Lundy et al. 1999; Baus et al. 2005; Kenchington et al. 2006). The dispersal ability is highly determined by the life history of a certain species, e.g., sessile and benthic species lacking larval stages usually show higher population genetic differentiation than those species with long planktonic larval stages (Hunt 1993; Kyle and Boulding 2000; Collin 2001). Marine currents are favorable to population mixing through long-distance dispersal of gametes, larvae, and adults, which dispel geographical differentiation (Riginos and Nachman 2001; Luttikhuizen et al. 2003; Kenchington et al. 2006).
Almost all marine bivalves have a long planktonic larvae stage. Passive larvae transport induced by marine currents is therefore expected to enhance gene flow and dispel geographical differentiation (Lewis and Thorpe 1994; Tremblay et al. 1994; Kyle and Boulding 2000; Fievet et al. 2006). Consequently, genetic analyses over either large or fine geographical scales failed to identify significant population structure in most marine bivalves, including mussels (Levinton and Koehn 1976; Skibinski et al. 1983), clams (Benzie and Williams 1992; Vadopalas et al. 2004), and scallop (Herbinger et al. 1998). Surprisingly, genetic differentiation over small geographical scales has only been recognized in a few studies (e.g., mussel Mytilus edulis, Ridgway 2001; clam Macoma balthica, Luttikhuizen et al. 2003). Some studies hypothesized that marine sessile bivalves differ from marine fishes and pelagic invertebrates in which the adults show a limited range of mobility (Baus et al. 2005; Kenchington et al. 2006). As a result, they tend to form discrete populations which display relatively unique community dynamics and population structure (Ridgway 2001; Luttikhuizen et al. 2003; Kenchington et al. 2006). However, for most marine bivalves including clams, scallops, mussels, and oysters, the mechanisms for driving geographical differentiation and creating the genetic population structure are still unknown.
Marine scallop, which comprises more than 300 species identified in worldwide oceans, is one important fauna of bivalve at both a commercial level and an ecological level. Consequently, numbers of aquaculture-related and ecology-concerned investigations were performed, such as marker-assisted selection (e.g., Qin et al. 2007; Zhan 2007), functional genomics (e.g., Song et al. 2006; Zhang et al. 2007, 2008a, b), planktonic larvae and species identification (e.g., Wang et al. 2006; Zhan et al. 2008), and phylogenetics (e.g., Wang et al. 2007). Additionally, some population genetics analyses tried to dissect the dynamics and mechanisms of diversification in marine scallop populations and to point out what are the main factors causing the population differentiation (e.g., Nagashima et al. 2005; Saavedra and Peña 2005; Kenchington et al. 2006). However, the results from different studies are mixed.
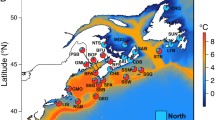
Zhikong scallop (Chlamys farreri Jones et Preston 1904), which belongs to Mollusca, Lamellibranchia, Pterimorphia, Pterioodae, Pectinidae, has a natural distribution along the seacoasts of China, Japan, Korea, and also Sakhalin of Russia. Like many marine bivalves, Zhikong scallop is sedentary as adult and has a long passive larval stage (about 15 days under natural conditions), large population size, and very high fecundity (usually more than 1 × 106 eggs per mature female). However, compared with other marine bivalves, Zhikong scallop possesses a relatively narrow geographical distribution range (Fig. 1). In China, the seacoasts around the Shandong Peninsula and the Liaodong Peninsula are its natural habitats (approximately from 35° N to 39° N, Fig. 1). Due to these special aspects of Zhikong scallop, a series of questions can be addressed: is there any geographical differentiation or unique genetic structure in Zhikong scallop populations at a fine geographical scale? If geographic differentiation or genetic structure is present, which factor(s) might drive this differentiation at such a fine geographical scale? The unusual oceanography around these two peninsulas, relative narrow distribution, and unique lifestyle of Zhikong scallop provide an interesting case for studies of local population genetic structure and integrative causes of geographical differentiation in marine bivalves at a fine scale. Additionally, as one of the most valuable fishery species in Eastern Asian countries, knowledge of the genetic structure and genetic differentiation of natural populations is also essential for aquaculture management and preservation of aquatic biodiversity for the sustainable development of scallop fisheries (Zhan 2007).

Among the molecular markers available for analysis of population genetic structure, microsatellite DNA markers are characterized as the finest identification of individuals and populations due to their hypervariability, abundance, neutrality, codominance, and unambiguous scoring of alleles (Tautz 1989; Weber and May 1989). In this study, a total of 270 individuals sampled from six populations were genotyped with nine microsatellite markers to clarify the population genetic diversity, to evaluate the genetic differentiation among geographical populations, to elucidate the impact of marine currents on the genetic structure and geographical differentiation, and to discuss the causes of genetic differentiation of marine bivalve species at fine geographical scales.
Materials and Methods
Sample Collection
Sample sites were chosen to cover all portions of the species range of its main natural habitats in the Northern China. Based on the biogeography and physical oceanography, there are six demographically independent populations with relatively large sizes in this narrow region, of which five are appropriately located inside four different marine retention gyres and one is separated by a marine channel (Laotieshan Channel) with very strong torrential currents (Fig. 1).
Six local populations were sampled from two different seas. One sample site was chosen in the Bohai Sea: the seacoast around the Southern Changshan Islands in Changdao County; one site was located in the boundary of the Bohai Sea and the Yellow Sea: the Taozi Bay in Yantai City. These two populations were located in the same marine gyre (Fig. 1). Four other sample localities were in the Yellow Sea: the seacoast around the Zhangzi Island in Dalian City, the Sanggou Bay in Rongcheng City, the Lingshan Bay in Jiaonan City, and the seacoast around the Qiansan Islands in Rizhao City. These four populations were in different marine gyres or separated by a marine channel (Laotieshan Channel; Table 1, Fig. 1). A total of 270 individuals were collected and shipped to our laboratory. The adductor muscle was removed from each living scallop and kept in a −80°C refrigerator prior to genetic analysis performed.
DNA Extraction and PCR Amplification
DNA was extracted from the frozen adductor muscles according to the protocol described by Zhang et al. (2004) with some minor modifications. Briefly, the muscle of each sample was homogenized in TNES buffer (10 mM Tris–HCl, pH 7.4; 10 mM ethylenediaminetetraacetic acid, pH 8.0; 125 mM NaCl; 0.5% sodium dodecyl sulfate and 4 M urea) and treated with 200 μg proteinase K (Merck) at 56°C overnight. The reaction mix was extracted with phenol–chloroform–isopentanol (25:24:1) and then precipitated with isopropanol and dissolved in 0.1× TE buffer (pH 8.0).
The population structure was investigated with nine polymorphic microsatellite markers, seven from Zhan et al. (2006a; CFMSP003, CFMSP007, CFMSP011, CFMSP075, CFMSM009, CFMSM014, and CFMSM020), one from Zhan et al. (2007a; CFBD018), and the remaining one from Zhan (2007; CFSSR001). The polymerase chain reactions (PCRs) were performed in a thermal cycler (GeneAmp PCR System 9700, Perkin-Elmer ABI, Co. Ltd.) in 15-μL mix containing 1× PCR buffer (Takara), 0.5 U Taq polymerase (Takara), 1.5 mM of Mg2+, 200 μM of each dNTP, 0.2 μM of each primer, and 40 ng of genomic DNA. The PCR program for all loci was set at: 5 min for an initial denaturation; 95°C for 30 s, locus-specific annealing temperature for 30 s, 72°C for 45 s repeated 35 times and ending with 72°C for 5 min.
DHPLC Analysis for Individual Genotyping
PCR products were separated on an automatic denaturing high-performance liquid chromatography (DHPLC) equipment: 3500 HT WAVE DNA fragment analysis system (Transgenomic Inc., USA). The amplified DNA fragments were loaded on a C18 reverse-phase column based on alkylated polystyrene divinylbenzene particles (DNASep™ column). The column temperature was set at 50°C for DNA fragment size separation. The gradient parameters were set as: minimum size 50 bp, maximum size 400 bp, and minutes per 100 bp 4 min. The cloned alleles (if available) and pUC19–HaeIII were separated as the molecular standards. The peaks representing different alleles were distinguished by the retention time, and the allele was named by its length (bp).
Data Analysis
For each locus in each population, the standard indices, including the number of alleles (A), allelic and genotypic frequencies, and observed heterozygosity (H o) and expected heterozygosity (H e) were calculated by POPGENE (Raymond and Rousset 1995) online version (http://wbiomed.curtin.edu.au/genepop/) or ARLEQUIN (Schneider et al. 1997) version 3.1 software package. The size (S c) and frequency (F c) of the most common allele and the size (S u) and frequency (F u) of unique alleles for a certain population were determined at each locus in all six populations. An index, the allelic richness (R s; El Mousadik and Petit 1996), which is suitable for comparison of the mean number of alleles among populations irrespective of sample size, was calculated by the software Fstat (Goudet 2001) version 2.9.3. The Hardy–Weinberg equilibrium (HWE) for each locus in each population was performed using the Markov chain randomization test (Guo and Thompson 1992) to estimate the exact P values. The significant criteria were adjusted for the sequential Bonferroni correction (Rice 1989).
Differences among populations in their genetic composition were examined in several ways. The index of pairwise F ST (θ estimator) based on an infinite alleles model (IAM) was estimated to assess the magnitude of differentiation among geographical populations, and an unbiased estimate of the significance of the probability test was calculated through 1,000 iterations employed by ARLEQUIN software. Additionally, R ST values based on a stepwise mutation model (SMM) were calculated using the program RstCalc (Goodman 1997). A log-likelihood G test (Goudet et al. 1996) employed by GENEPOP software was used to determine whether or not the allelic and genotypic distribution was identical between all pairs of populations. The significance of the P values across all the loci for all populations was determined using Fisher’s probability combination test, and significance in pairwise comparison was evaluated after a sequential Bonferroni adjustment of critical probabilities.
The relationship among geographical populations was estimated from Nei’s standard genetic distances (D A) between all pairs of populations (Nei 1987), and the distance matrices were used to construct neighbor-joining (NJ) dendrogram. The bootstrap values for this tree were calculated using 1,000 replicates by resampling microsatellite loci. All calculations were conducted using the PHYLIP 3.6 software package (Felsenstein 2000).
We used the assignment test and Monmonier algorithm test to clarify the geographical differentiation. Assignment methods have proven to be useful tools in detecting the influence of marine currents on population genetic structure (Fievet et al. 2006). The likelihood of an individual originating from a given population was estimated by using a Bayesian-based method implemented in the program GeneClass2 (Piry et al. 2004). The assignment threshold score was set as 0.05. Due to sampling from the same marine gyre and no detectable differentiation with CD population (see “Results”), YT population, which was sampled from the boundary of the Bohai Sea and the Yellow Sea, was assigned to the CD–YT group in the Bohai Sea when the pooled data were calculated. The latitude and longitude coordinates were provided for each individual and used along with the genetic data collected from the microsatellite analysis to generate a connectivity network of genetic distances based on Delaunay triangulation using Barrier software version 2.2 (Manni et al. 2004). Monmonier’s maximum difference algorithm was then used to identify putative genetic boundaries across the oceanographic landscapes.
Results
Genetic Diversity of Different Populations
Measures of genetic diversity for each population were calculated from the observed allele distribution and are presented in Table 2. A total of 144 alleles were detected across nine microsatellite loci ranging from two alleles at the locus CFMSM009 in the JN population to 22 alleles at the locus CFMSP003 in the RZ population. The total number of alleles per locus ranged from 5.7 to 18.5, and the total number of alleles per population ranged from 9.0 to 14.6, respectively. Over all microsatellite loci, the RZ population had the greatest average number of alleles, while the least number of alleles was detected in the JN population (Table 2). The allelic richness ranged from 8.2 to 13.4, and populations RZ and JN showed the highest and lowest values, respectively. The number of alleles observed at a certain locus varied highly among populations, e.g., only two alleles were detected at the locus CFMSM009 in the JN population, while much higher values were observed at this locus in the other populations (Table 2).
The high within-population diversity was also reflected in the levels of the expected and observed heterozygosities, which varied from 0.5135 to 0.6604 and from 0.6632 to 0.8173, respectively. On average, the RC population had the highest H e value (0.8173) and the JN population showed the lowest (0.6632; Table 2). Considering the sea area, the average H e of the populations located in the Bohai Sea was lower than those sampled from the Yellow Sea (0.7136 vs 0.7415).
A total of 17 alleles were found to be unique to a single population (Table 2). Our unique allele analysis of samples pooled by different seas suggested that populations sampled from the Bohai Sea had fewer unique alleles. No unique allele was detected in the YT population and only one was found in the CD population, while the higher number of unique allele was observed in the other four populations sampled from the Yellow Sea (Table 2). Interestingly, the RZ population displayed a large number of unique alleles at the loci CFBD018 and CFMSM014 (Table 2). The analysis of the most frequent allele showed that only one locus (CFSSR001) exhibited a single most frequent allele (196 bp) in all populations; however, variable numbers were observed at the other loci among six populations (Table 2). While we realize that the sample size could be a factor to cause changes in the number of unique alleles or low frequency alleles, it should not affect the most common allele and its frequency. The different common alleles among populations strongly suggest genetic differentiation.
Forty cases of observed genotype distribution were generally in accord with Hardy–Weinberg proportion; however, 14 cases remained significant after adjustment of P values with the sequential Bonferroni method, and these departures were due to an excess of homozygotes (Table 2). No locus or sampling site had a particularly high number of sites–loci that were deviated from HWE, and no heterozygosity excess was detected in any of the populations at any locus.
Genetic Differentiation Among Different Populations
The overall genetic differentiation among populations was substantial with both IAM- and SMM-based estimators. When pairwise multilocus F ST and R ST analyses were performed, the populations sampled from the Yellow Sea showed significant differentiation from each other. The populations from the Yellow Sea also differed from two populations: the CD population sampled from the Bohai Sea and the YT population collected from the boundary of the Bohai Sea and the Yellow Sea. However, no significant difference was detected between populations CD and YT (Table 3).
Genetic differentiation among geographical populations was also observed when pairwise comparisons of allelic and genotypic frequencies were performed. The comparison revealed that, of 135 pairs of locus–population cases, 116 and 122 cases were significantly different in allelic and genotypic frequencies, respectively, after sequential Bonferroni corrections. All loci in populations RC, JN, and RZ were significantly different in all pairs of both allelic and genotypic frequencies. The DL population differed from the CD population at five loci in allelic frequencies but at six loci in genotypic frequencies, while the DL population differentiated from the YT population at five loci in allelic frequencies and at seven loci in genotypic frequencies, respectively. The differentiation was only observed at one locus in both allelic and genotypic frequencies between the CD population and the YT population.
D A distances between populations were estimated using the data from nine microsatellite loci, and a tree was constructed using the NJ method (Fig. 2). Neighbor-joining trees were also constructed using pairwise F ST and R ST distances, and these trees gave the same topology. The trees also demonstrate separate clustering of populations, which further illustrate the genetic differentiation.

Neighbor-joining tree topology showing the genetic relationship among six populations of Zhikong scallop (C. farreri)
Individual Assignment
Provided that populations CD and YT are considered as one group, 95.6% of the individuals were assigned to their sampling populations/group. Based on the multilocus genotyping, populations RC, JN, and RZ showed a complete match of all individuals to their sampling populations. For the DL population and the CD–YT group, a total of 12 individuals were excluded and alternatively assigned to the other populations with the probabilities varying from 71.2% to 98.6%.
Genetic Barrier Predication
Genetic boundaries estimated from microsatellite DNA data are presented in Fig. 3. Surprisingly, the first two barriers (A and B) did not separate the two different seas, the Bohai Sea and the Yellow Sea into two parts, but split the Yellow Sea into two regions and separated populations JN and RZ from other populations. The third boundary (C) was predicted to isolate the RC population in process, and the last boundary (D) isolated the DL population, which also separated the Yellow Sea and the Bohai Sea (Figs. 1 and 3). The patterns of genetic barriers predicted by the Barrier software were highly consistent with all the statistical analyses such as NJ tree, results of pairwise F ST, and, most importantly, with the marine gyres (Figs. 1 and 3). All these results stated above support the hypothesis that marine currents drive the geographical differentiation and generate the population genetic structure.

Genetic barriers predicted by software BARRIER (version 2.2). Delaunay triangulation (thin lines) and genetic boundaries (bold lines) obtained using Monmonier’s maximum difference algorithm are shown
Discussion
The hypothesis that the high dispersal capacities of marine bivalves with extended larval phases should lead to wide spread over large geographical scales, and the interaction between the biological characteristics and marine currents may lead to poor geographical differentiation (e.g., Arnaud et al. 2000). This hypothesis has been evidenced by some studies in bivalve population differentiation (e.g., Skibinski et al. 1983; Vadopalas et al. 2004); however, some studies were against this popular hypothesis (e.g., Ridgway 2001; Luttikhuizen et al. 2003). Actually, there are few direct experimental evidences in most of the marine bivalves until now, and few studies are concerned on the relationship between genetic structure or geographical differentiation and marine landscapes. One way to obtain further insight into the oceanography factors which create the genetic structure of populations is to combine genetic data with information on landscape characteristics to study how landscape structures impact population structure (Fievet et al. 2006; Jørgensen et al. 2005; Kenchington et al. 2006). The use of landscape genetic approaches to elucidate genetic barriers and to test the membership of individuals has proven very useful in this study to explain the geographical differentiation. For Zhikong scallop, the results in this study show there is good agreement between hydrographical barriers and genetic differentiation of geographical populations.
High Population Diversity and Deviation from HWE
A high level of genetic diversity was detected in all six populations. Two leading indices, allelic diversity and heterozygosity, are considerably high (Table 2). High genetic diversity seems to be one common characteristic of marine bivalves, such as oysters (e.g., Crassostrea gigas, Li et al. 2003; Crassostrea virginica, Brown et al. 2000; Pinctada maxima, Smith et al. 2003), clams (e.g., Panopea abrupta, Vadopalas et al. 2004; Scapharca broughtonii, An and Park 2005), and scallops (e.g., Placopecten magellanicus, Kenchington et al. 2006; Argopecten irradians, Zhan et al. 2005, 2006b). The large population size and high nucleotide mutation rate may be the main contributing factors (Callen et al. 1993; Launey and Hedgecock 200l; Hedgecock et al. 2004; Zhan et al. 2007b).
Most of the cases were in HWE; however, 14 locus–population cases showed significant deviation from HWE. The tests showed that these deviations were due to a heterozygote deficiency. Significant heterozygote deficiency has been commonly reported in marine invertebrate species, and null alleles, frequently found at microsatellite DNA loci, are potential causes for heterozygote deficiency in HWE tests (Callen et al. 1993; Li et al. 2006). For marine bivalves, the importance of null alleles as an explanation for heterozygote deficiency was also considered and further discussed in the studies of population genetic diversity, inheritance pattern of microsatellites, and construction of the SSR-based linkage maps (e.g., Li et al. 2003; Vadopalas et al. 2004; Hubert and Hedgecock 2004; Baranski et al. 2006; Zhan et al. 2007b; Zhan 2007). In fact, null alleles have been evidenced at some of these loci in construction of SSR linkage maps for the Zhikong scallop (Zhan 2007). The wide spread of null allele is the other characteristic of marine bivalves, e.g., about 55.2% of loci had null allele(s) from two controlled crosses established for construction of SSR linkage maps in the Zhikong scallop (Zhan 2007). Li et al. (2003) also reported a comparatively high ratio (51.9%) of null alleles in the Pacific oyster (C. gigas). The prevalent facts confirmed by experimental evidences that a high percentage of primers containing variable nucleotides may be responsible for the widespread appearance of null alleles in bivalves (Li et al. 2003; Hedgecock et al. 2004; Zhan et al. 2007b). We should be aware that low heterozygosity estimates in a population and deviation from HWE at a certain locus might be partially due to the null allele(s) in the study of population genetic diversity.
Impact of Marine Gyres/Currents on Population Differentiation
All the statistical results, including the unique allele and most common allele analyses (Table 2) and allelic and genotypic frequency comparisons, show that populations CD and YT showed no significant differentiation. The low F ST values and result of individual assignment also strongly suggested the high gene flow between these two populations. It is well known that the Bohai Sea is a semiclosed sea with relatively poor water exchange with its neighboring sea. Most importantly, these two populations were sampled from the same marine gyre (Fig. 1). The passive larvae transport induced by the intercirculated currents may prevent the differentiation between these two populations. The population mechanism was also assessed by Tremblay et al. (1994) when they modeled the distribution of scallop larvae. They observed that the scallop larvae in the vicinity of Georges (Canada) and Georges (USA) were strongly coupled and can mix in a single generation. These results suggest that the marine currents which cause the spread of planktonic larvae may be a key factor in explaining the weak differentiation in planktonic-developing species.
Several population studies and phylogeographical investigation in marine organisms have also shown that the population structure of species with planktonic larval development (i.e., planktotrophic development species) are also more sensitive to marine currents than those with direct development (i.e., direct development species; e.g., Show et al. 1999; Kyle and Boulding 2000; Baus et al. 2005; Fievet et al. 2006). The planktonic period usually provides a large capacity to disperse widely over the oceans, which therefore results in spatial homogeneity of their genetic structure (Avise 2004). Comparatively, larvae were suspected to migrate little from one zone to another or migrate in a single direction due to local currents (e.g., scallop Aequipecten opercularis, Lewis and Thorpe 1994; giant clam Tridacna derasa, Macaranas et al. 1992) and hydrographical barriers (e.g., Palumbi and Baker 1994).
Estimate of differentiation was expected to be low because of the high variability of microsatellite loci (Hedrick 1999); however, significant differentiation was observed among Zhikong scallop populations sampled from a fine geographical scale. Significant structure was found at a large geographical scale in the studies of differentiation of marine bivalves (e.g., Arnaud et al. 2000; Launey et al. 2002). However, few studies have shown significant genetic differentiation over a fine geographical scale in marine bivalves. In the present study, three populations (RC, JN, and RZ) sampled from the Yellow Sea showed high pairwise F ST and R ST values, indicating that these three populations are genetically divergent. The landscape genetic analysis showed significant genetic boundaries between these populations, and these barriers were consistent with the marine gyres/currents in the sampling localities (Figs. 1 and 3). Additionally, results of assignment tests revealed that a population rarely exchanged migrants with other populations located within the area defined by this boundary. Besides the genetic differentiation detected by molecular methods, the RZ population showed some unique morphological characteristics, such as shell color, shell shape, and the number of thorns on the upper shells. Although the distance between populations JN and RZ is only 92 km, surprisingly, these two populations showed very different genetic makeup and morphological characteristics. Interestingly, no genetic differentiation was observed in the population pair (CD and YT) sampled from the same marine gyre with the comparative geographical distance (80 km). Based on the analyses in genetic barrier prediction and individual assignment, the marine currents can be responsible for the surprise difference between these two cases.
As described for numerous marine species, the Laotieshan Channel (Fig. 1) may represent a major barrier to the dispersal for Zhikong scallop. Migration (gene flow) could be restricted by marine currents, in particular the strong currents occurring in the narrow passage of the Laotieshan Channel. In the present study, the putative genetic barrier, result of individual assignment, and differentiation analyses were basically consistent with this geographical characteristic. However, six individuals derived from the DL population were excluded and assigned to the CD–YT group, while six specimens of the CD–YT group were similarly rejected and assigned to the DL population. With the development of scallop aquaculture, the areas around the Zhangzi Island in Liaoning Province and the Taozi Bay in Shandong Province become the most important distributing centers for scallop seedlings and adults in China. The scallop aquaculture activities may introduce larvae/adults into new geographical locations. The factor for larvae/adults’ introduction cannot be ruled out without further investigation for the explanation of the individual assignment results. We should be aware that, if a large number of invaded scallops are released or mixed to the natural environment by anthropogenic activities, they could alter the genetic composition of wild populations by either displacing them or interbreeding with them (Norris et al. 1999). Therefore, proper management of farmed populations and the assessment of risks caused by released or escaped individuals should be fully considered in modern scallop aquaculture.
The genetic differentiation of marine bivalves sampled over a large scale in some studies were consistent with a model of isolation by distance (IBD; e.g., pearl oyster, Arnaud et al. 2000; European flat oyster, Launey et al. 2002; Rose et al. 2006), where the populations are connected by continuous migration but the rate of gene flow is greatest between neighboring populations. However, the analyses including the genetic barrier prediction and individual assignment showed the habitat fragmentation formed by marine gyres/currents is responsible for the geographical differentiation in Zhikong scallop. Generally, the IBD model was considerably strengthened over a large geographical scale analysis, while habitat fragmentation is much competent for the explanation of differentiation for a fine geographical scale (e.g., Launey et al. 2002; Wilson et al. 1999). Due to the hydrographic complexity in the habitats of Zhikong scallop, continuous migration may be interrupted by the bays, marine gyres and currents, peninsulas, and/or other still unknown integrative physical barriers; therefore, we should be aware that it might be too simplistic to describe the population differentiation by the simple models of panmixia, islands, bays, marine currents/gyres, and IBD.
Conclusion
The results of the present study suggest that the marine currents play one of the important roles in defining population differentiation. Our results evidenced for the first time that genetic differentiation of marine scallop was in relation to habitat fragmentation over a fine geographical scale separated by marine gyres/currents. Additionally, it should be pointed out that there are several difficulties in studying the geographical differentiation of marine bivalves at fine geographical scales, such as difficult sampling, limited population numbers, and lots of unknown marine environmental factors. Although only six populations were analyzed in this study, the sampling sites covered all the natural distribution range and almost all the dominant natural populations in the Northern China. More importantly, the landscape genetics analysis evidenced that the marine gyres/currents can be responsible for the geographical differentiation. Based on the unique issues stated above, at least three aspects should be recommended for a long-term population surveillance in future studies on the geographical differentiation in marine bivalves, (1) the models and mechanisms of the migration and dispersal, especially for germ cells, embryos, and larvae, (2) full consideration of more oceanographic factors for further landscape genetic analysis, such as marine currents, retention gyres associated with banks or embayments, and the distribution of rocky formations of sea beds for the eventual larvae settlement, and (3) anthropogenic activities associated with the aquaculture and fisheries.
References
An HY, Park JY (2005) Ten new highly polymorphic microsatellite loci in the blood clam Scapharca broughtonii. Mol Ecol Notes 5:896–898
Arnaud S, Monteforte M, Galtier N, Bonhomme F, Blanc F (2000) Population structure and genetic variability of pearl oyster Pinctada mazatlanica along Pacific coasts from Mexico to Panama. Conserv Genet 1:299–307
Avise JC (2004) Molecular markers, natural history, and evolution, 2nd edn. Sinauer, Sunderland
Baranski M, Loughnan M, Austin CM, Robinson N (2006) A microsatellite linkage map of the black lip abalone, Haliotis rubra. Anim Genet 37:563–570
Baus E, Darrock DJ, Bruford MW (2005) Gene-flow pattern in Atlantic and Mediterranean populations of the Lusitanian sea star Asterina gibbosa. Mol Ecol 14:3373–3382
Benzie JAH, Williams ST (1992) No genetic differentiation of giant clam (Tridacna gigas) populations in the Great Barrier Reef, Australia. Mar Biol 113:373–377
Bohonak AJ (1999) Dispersal, gene flow, and population structure. Quart Rev Biol 74:21–45
Brown B, Franklin DE, Gaffney PM, Hong M, Dendanto D, Kornfield I (2000) Characterization of microsatellite loci in the eastern oyster, Crassostrea virginica. Mol Ecol 9:2217–2219
Callen DF, Thompson AD, Shenm Y, Phillipsm HA, Richards RI, Mulley JC, Sutherland GR (1993) Incidence and origin of “null” alleles in the (AC)n microsatellite markers. Am J Hum Genet 52:922–927
Collin R (2001) The effects of mode of development on phylogeography and population structure of North Atlantic crepidula (Gasteropoda: Calyptraeidae). Mol Ecol 10:2249–2262
Edmands S, Potts DC (1997) Population genetic structure in brooding sea anemones (Epiactis spp.) with contrasting reproductive modes. Mar Biol 127:485–498
El Mousadik A, Petit RJ (1996) High level of genetic differentiation for allelic richness among populations of the argan tree (Argania spinosa (L.) Skeels) endemic to Morocco. Theor Appl Genet 92:832–839
Felsenstein J (2000) PHYLIP version 3.6. Department of Genetics, University of Washington, Seattle
Fievet V, Touzet P, Arnaud J, Cuguen J (2006) Spatial analysis of nuclear and cytoplasmic DNA diversity in wild sea beet (Beta vulgaris ssp. maritima) populations: do marine currents shape the genetic structure? Mol Ecol 16:1847–1864
Goodman SJ (1997) Rst Calc: a collection of computer programs for calculating estimates of genetic differentiation from microsatellite data and determining their significance. Mol Ecol 6:881–885
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). http://www.unil.ch/izea/softwares/fstat.html
Goudet J, Raymond M, De Meiis T, Rousset F (1996) Testing differentiation in diploid populations. Genetics 144:933–940
Guo SW, Thompson EA (1992) Performing the exact test of Hardy–Weinberg proportion for multiple alleles. Biometrics 48:361–372
Hedgecock D, Li G, Hubert S, Bucklin K, Ribes V (2004) Widespread null alleles and poor cross-species amplification of microsatellite DNA loci cloned from the Pacific oyster, Crassostrea gigas. J Shellfish Res 23:379–385
Herbinger CM, Vercaemer BM, Gjetvaj B, O’Dor R (1998) Absence of genetic differentiation among geographically close sea scallop (Placopecten magellanicus) beds with cDNA and microsatellite markers. J Shellfish Res 17:117–122
Hedrick PW (1999) Genetics of populations, 2nd edn. Jones and Bartlett, Sudbury, p 629
Hubert S, Hedgecock D (2004) Linkage maps of microsatellite DNA markers for the Pacific oyster Crassostrea gigas. Genetics 168:351–362
Hunt A (1993) Effects of contrasting patterns of larval dispersal on the genetic connectedness of local populations of two intertidal starfish, Patiriella calcar and P. exigua. Mar Ecol Prog Ser 92:179–186
Jørgensen HBH, Hansen MM, Bekkevold D, Ruzzante DE, Loeschcke V (2005) Marine landscapes and population genetic structure of herring (Clupea harengus L.) in the Baltic Sea. Mol Ecol 14:3219–3234
Kenchington EL, Patwary MU, Zouros E, Bird CJ (2006) Genetic differentiation in relation to marine landscape in a broadcast-spawning bivalve mollusc (Placopecten magellanicus). Mol Ecol 15:1781–1796
Kyle CJ, Boulding EG (2000) Comparative population genetic structure of marine gastropods (Littorina spp.) with and without pelagic larval dispersal. Mar Biol 137:835–845
Launey S, Hedgecock D (2001) High genetic load in the Pacific oyster Crassostrea gigas. Genetics 159:255–265
Launey S, Ledu C, Boudry P, Bonhomme F, Naciri-Graven Y (2002) Geographic structure in the European flat oyster (Ostrea edulis L.) as revealed by microsatellite polymorphism. J Hered 93:331–351
Levinton JS, Koehn RK (1976) Population genetics of mussels. In: Bayne BL (ed) Marine mussels, their ecology and physiology. Cambridge University Press, Cambridge, pp 357–384
Lewis RI, Thorpe JP (1994) Temporal stability of gene frequencies within genetically heterogeneous populations of queen scallop Aequipecten (Chlamys) opercularis. Mar Biol 121:117–126
Li FH, Liu AJ (1989) Hydrological regime in the offshore regions of Shandong Peninsula. Shandong Maps Press, Shandong
Li G, Hubert S, Bucklin K, Ribes V, Hedgecock D (2003) Characterization of 79 microsatellite DNA markers in the Pacific oysters Crassostrea gigas. Mol Ecol Notes 3:228–232
Li Q, Yu H, Yu RH (2006) Genetic variability assessed by microsatellites in cultured populations of the Pacific oyster (Crassostrea gigas) in China. Aquaculture 259:95–102
Lundy CJ, Moran P, Rico C, Milner RS, Hewitt GM (1999) Macrogeographical population differentiation in oceanic environments: a case study of European hake (Merluccius merluccius), a commercially important fish. Mol Ecol 81:889–1898
Luttikhuizen PC, Drent J, Baker AJ (2003) Disjunct distribution of highly diverged mitochondrial lineage clade and population subdivision in a marine bivalve with pelagic larval dispersal. Mol Ecol 12:2215–2229
Macaranas JM, Ablan CA, Pante MJR, Benzie JAH, Williams ST (1992) Genetic structure of giant clam (Tridacna derasa) populations from reefs in the Indo-Pacific. Mar Biol 113:231–238
Manni F, Guérard E, Heyer E (2004) Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by “Monmonier’s algorithm”. Hum Biol 76:173–190
Nagashima K, Sato M, Kawamata K, Nakamura A, Ohta T (2005) Genetic structure of Japanese scallop population in Hokkaido, analyzed by mitochondrial haplotype distribution. Mar Biotechnol 7:1–10
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Nielsen EE, Kenchington E (2001) Prioritising marine fish and shellfish populations for conservation: a useful concept? Fish Fisher 2:328–343
Norris AT, Bradley DG, Cunningham EP (1999) Microsatellite genetic variation between and within farmed and wild Atlantic salmon (Salmo salar) populations. Aquaculture 180:247–264
Palumbi SR (1992) Marine speciation on a small planet. Trends Ecol Evol 7:114–118
Palumbi SR, Baker CS (1994) Contrasting population structure from nuclear intron sequences and mtDNA of humpback whales. Mol Biol Evol 11:426–435
Piry S, Alapetite A, Cornuet J-M, Paetkau D, Baudouin L, Estoup A (2004) geneclass2: a software for genetic assignment and first-generation migrant detection. J Hered 95:536–539
Qin Y, Liu X, Zhang H, Zhang G, Guo X (2007) Identification and mapping of amplified fragment length polymorphism markers linked to shell color in bay scallop, Argopecten irradians irradians (Lamarck, 1819). Mar Biotechnol 9:66–73
Raymond M, Rousset F (1995) Genepop (version 1.2): population genetic software for exact tests and ecumenicism. J Hered 86:248–249
Rice RW (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Ridgway G (2001) Interpopulation variation in blue mussels, Mytilus edulis L., over short distances. Sarsia 86:157–161
Riginos C, Nachman MW (2001) Population subdivision in marine environments: the contribution of biogeography, geographical distance and discontinuous habitat to genetic differentiation in a blennioid fish, Axoclinus nigricaudus. Mol Ecol 10:1439–1453
Rose CG, Paynter KT, Hare MP (2006) Isolation by distance in the Eastern oyster, Crassostrea virginica, in Chesapeake Bay. J Hered 97:158–170
Saavedra C, Peña JB (2005) Nucleotide diversity and Pleistocene population expansion in Atlantic and Mediterranean scallops (Pecten maximus and P. jacobaeus) as revealed by the mitochondrial 16S ribosomal RNA gene. J Exp Mar Biol Ecol 323:138–150
Schneider S, Kueffer JM, Roessi D, Excoffier L (1997) AREQUIN version 1.1: a software for population genetic data analysis. Genetics and Biometry Laboratory, University of Geneva, Geneva
Show PW, Pierce GJ, Boyle PR (1999) Subtle population structuring within a highly vagile marine invertebrate, the veined squid Loligo forbesi, demonstrated with microsatellite DNA markers. Mol Ecol 8:407–417
Skibinski DOF, Beardmore JA, Cross TF (1983) Aspects of the population genetics of Mytilus (Mytilidae; Mollusca) in the British Isles. Biol J Linn Soc 19:137–183
Smith C, Benzie JAH, Wolson KJ (2003) Isolation and characterization of eight microsatellite loci from silver-lipped pearl oyster Pinctada maxima. Mol Ecol Notes 3:125–127
Song L, Xu W, Li C, Li H, Wu L, Xiang J, Guo X (2006) Development of expressed sequence tags from the bay scallop, Argopecten irradians irradians. Mar Biotechnol 8:161–169
Tautz D (1989) Hypervariability of simple sequences as a general source for polymorphic DNA markers. Nucleic Acids Res 17:6463–6471
Tremblay MJ, Loder JW, Werner FE, Naimie CE, Page FH, Sinclair MM (1994) Drift of sea scallop larvae Placopecten magellanicus on Georges Bank: a model study of the roles of mean advection, larval behaviour and larval origin. Deep Sea Res 41:7–29
Vadopalas B, Leclair LL, Bentzen P (2004) Microsatellite and allozyme analyses reveal few genetic differences among spatially distinct aggregations of geoduck clams (Panopea abrupta, Conrad 1849). J Shellfish Res 23:693–706
Wang S, Bao ZM, Zhang LL, Li N, Zhan AB, Guo WB, Wang XL Hu JJ (2006) A new strategy for species identification of planktonic larvae: PCR–RFLP analysis of the internal transcribed spacer region of ribosomal DNA detected by agarose gel electrophoresis or DHPLC. J Plankton Res 28:375–384
Wang S, Bao ZM, Li N, Zhang LL, Hu JJ (2007) Analysis of the secondary structure of ITS1 in Pectinidae: implications for phylogenetic reconstruction and structural evolution. Mar Biotechnol 9:231–242
Weber JL, May PE (1989) Abundant class of human DNA polymorphism which can be typed using the polymerase chain reaction. Am J Hum Genet 44:388–396
Wilson AB, Naish K-A, Boulding EG (1999) Multiple dispersal strategies of the invasive quagga mussel (Dreissena bugensis) as revealed by microsatellite analysis. Can J Fish Aquat Sci 56:2248–2257
Zhan AB (2007) Development, characterization and application of microsatellite markers in Zhikong scallop (Chlamys farreri). Doctoral Dissertation of Ocean University of China, Qingdao
Zhan AB, Bao ZM, Wang XL, Hu JJ (2005) Microsatellite markers derived from bay scallop Argopecten irradians expressed sequence tags. Fisheries Sci 71:1341–1346
Zhan AB, Bao ZM, Yao B, Wang XL, Min H, Hu JJ (2006a) Polymorphic microsatellite markers in the Zhikong scallop Chlamys farreri. Mol Ecol Notes 6:127–129
Zhan AB, Hu JJ, Wang XL, Lu W, Hui M, Bao ZM (2006b) A panel of polymorphic EST-derived microsatellite loci for the bay scallop (Argopecten irradians). J Mollus Stud 72:436–438
Zhan AB, Bao ZM, Hu XL, Hui M, Wang ML, Peng W, Zhao HB, Hu JJ (2007a) Isolation and characterization of 150 novel microsatellite markers for Zhikong scallop (Chlamys farreri). Mol Ecol Notes 7:1015–1022
Zhan AB, Bao ZM, Hui M, Wang ML, Zhao HB, Lu W, Hu XL, Hu JJ (2007b) Inheritance pattern of EST-SSRs in self-fertilized larvae of the bay scallop Argopecten irradians. Ann Zoo Fenn 44:259–268
Zhan AB, Bao ZM, Hu XL, Lu W, Wang S, Peng W, Wang ML, Hui M, Hu JJ (2008) Accurate methods of DNA extraction and PCR-based genotyping for single scallop embryos/larvae long preserved in ethanol. Mol Ecol Resources 8:790–795
Zhang QQ, Xu XF, Qi J, Wang XL, Bao ZM (2004) The genetic diversity of wild and farmed Japanese flounder populations. Period Ocean Univ China 34:816–820
Zhang LL, Bao ZM, Cheng J, Li H, Huang XT, Wang S, Zhang C, Hu JJ (2007) Fosmid library construction and initial analysis of end sequences in Zhikong scallop (Chlamys farreri). Mar Biotechnol 9:606–612
Zhang LL, Bao ZM, Wang S, Hu XL, Hu JJ (2008a) FISH mapping and identification of Zhikong scallop (Chlamys farreri) chromosomes. Mar Biotechnol 10:151–157
Zhang Y, Zhang X, Scheuring CF, Zhang HB, Huan P, Li F, Xiang J (2008b) Construction and characterization of two bacterial artificial chromosome libraries of Zhikong Scallop, Chlamys farreri Jones et Preston, and identification of BAC clones containing the genes involved in its innate immune system. Mar Biotechnol 10:358–365
Zhou D, Liang YB, Tseng CK (1994) Oceanology of China seas. Kluwer Academic, Boston
Acknowledgements
We wish to acknowledge numerous researchers who help collect the live scallops and provide the oceanography information. Great thanks to Drs. Jinzhong Fu, Hyuk Je Lee, Elizabeth G. Boulding, and the other members in the discussion group of evolution and ecology biology for commenting on the early versions of this manuscript, Daniel Noble for English language correction, Yan Li for artwork assistance, and anonymous reviewers for extensive and constructive comments. This research was funded by The National High Technology Research and Development Program of China (“863” program, 2006AA10A408), Specialized Research Fund for the Doctoral Program of Higher Education (20060423015), National Key Technology Research and Development Program of China (2006BAD09A09, 2006BAD09A10) and NCET-06-0594.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhan, A., Hu, J., Hu, X. et al. Fine-Scale Population Genetic Structure of Zhikong Scallop (Chlamys farreri): Do Local Marine Currents Drive Geographical Differentiation?. Mar Biotechnol 11, 223–235 (2009). https://doi.org/10.1007/s10126-008-9138-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-008-9138-1