
Abstract
Hyperimmunoglobulin D syndrome (HIDS) is a hereditary autoinflammatory disease characterized by recurrent inflammatory attacks with fever, abdominal pain, lymphadenopathy, aphthous stomatitis, and skin lesions. There are few reports on HIDS patients complicated with macrophage activation syndrome (MAS); however, to our knowledge, there is no case of HIDS with recurrent MAS attacks. We report two pediatric patients initially diagnosed as Kawasaki disease and systemic juvenile idiopathic arthritis presented with recurrent MAS episodes with prolonged fever, skin rash, hepatosplenomegaly, cervical lymphadenopathy, aphthous stomatitis, headache, pancytopenia, hyperferritinemia, and hypofibrinogenemia, finally diagnosed as HIDS with a documented homozygous MVK gene mutation. This is the first report on recurrent MAS attacks due to HIDS in pediatric patients who were successful treated with corticosteroids and anti-IL-1 therapies. Thus, clinicians should be vigilantly investigated signs of autoinflammatory diseases in patients with recurrent MAS attacks during their disease course, and HIDS should be considered an underlying disease for triggering recurrent MAS attacks. We have also reviewed the current literature regarding HIDS cases complicated with a MAS attack and summarized their demographic, treatment, and outcome characteristics.
Key points • Hyperimmunoglobulin D syndrome should be considered in differential diagnosis in patients who experienced recurrent macrophage activation syndrome attacks. |
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mevalonate kinase deficiency (MKD) is an autosomal recessive inherited autoinflammatory disorder affecting various organs and has two distinct phenotypes; the severe form is called as mevalonic aciduria (MVA), and milder form as hyperimmunoglobulin D syndrome (HIDS) [1]. MKD is caused by mevalonate kinase (MVK) gene mutation resulting in deficiency of mevalonate kinase enzyme, which plays a role in cholesterol and non-sterol isoprenoid biosynthesis [2]. HIDS is characterized by recurrent attacks of fever, maculo-papular skin rash, cervical lymphadenopathy, abdominal pain, headache, and aphthous stomatitis lasting 3 to 6 days [1]. Clinical symptoms of HIDS typically begin in infancy period and febrile attacks can be triggered by vaccination, surgery, trauma, or stress. Urine mevalonic acid, serum acute phase reactants, and immunoglobulin D levels are usually elevated in inflammatory attacks [3, 4]. Non-steroidal anti-inflammatory drugs (NSAIDs) and short-term corticosteroids are useful treatments to reduce symptoms especially during inflammatory attacks [4, 5]. Anti-interleukin-1 (IL-1) therapies have shown a beneficial effect to impede the inflammatory flares [6]. There are also some cases with HIDS responsive to anti-tumor necrosis factor (TNF) therapy [7]. Amyloidosis is the most important devastating complication in undiagnosed and/or untreated patients with HIDS [8].
Macrophage activation syndrome (MAS) is a life-threatening condition due to excessive production of cytokines with aberrant activation of T lymphocytes and macrophages, characterized by persistent fever, cytopenia, hyperferritinemia, hypertriglyceridemia, hypofibrinogenemia, elevated liver function tests, coagulopathy, hepatosplenomegaly, encephalopathy, and multi-organ failure [9]. MAS may develop in patients with infectious, malignant, autoimmune, and autoinflammatory diseases, such as systemic juvenile idiopathic arthritis (sJIA), Kawasaki disease (KD), systemic lupus erythematosus, tumor necrosis factor receptor–associated periodic syndrome (TRAPS), deficiency of adenosine deaminase 2, and HIDS [10,11,12,13,14,15,16]. The main goal of treatment of MAS is to depress the hyperinflammation with corticosteroids, cyclosporine A (CSA), intravenous-immunoglobulin (IVIG), anti-IL-1 therapies and/or plasmapheresis [17]. Some patients may exhibit a recurrent disease course irrespective of treatment.
Herein, we report two pediatric patients presented with recurrent MAS episodes with prolonged fever, skin rash, hepatosplenomegaly, cervical lymphadenopathy, pancytopenia, hyperferritinemia, and hypofibrinogenemia, finally diagnosed with HIDS. To our knowledge, this is the first report on recurrent MAS attacks due to HIDS in pediatric patients who were successfully treated with corticosteroids and anti-IL-1 therapies.
Case presentations
Case 1
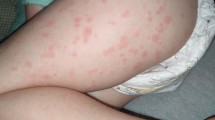
A 20-month-old girl was referred to the pediatric rheumatology clinic with prolonged fever, oral aphthous stomatitis, maculopapular skin rash, and headache lasting for 10 days. Physical examination showed maculopapular skin rash at the extremities and trunk, cracked lips, a 2-cm-diameter-sized unilateral cervical lymphadenopathy, hepatosplenomegaly, and neck stiffness. There was no parental consanguinity. Laboratory workup showed leukopenia, thrombocytopenia, hypofibrinogenemia, elevated acute phase reactants, and high ferritin and triglyceride levels. Anti-nuclear antibody (ANA), anti-dsDNA, and rheumatoid factor (RF) were negative, and serum complement levels, urinalysis, blood peripheral smear, and chest X-ray were normal. Viral serology and brucella tests were unremarkable. Abdomen ultrasonography (USG) revealed hepatosplenomegaly, while cervical USG revealed lymphadenopathy resembling reactive enlargement. Echocardiography findings, including coronary arteries, were normal. Cerebrospinal fluid analysis from lumbar puncture were normal. The diagnosis of incomplete KD was considered because of the presence of prolonged fever and three accompanying clinical findings, including cervical lymphadenopathy, mucosal involvement, and skin rash. Hemophagocytic macrophages were revealed in bone marrow aspiration. Our first decision was MAS secondary to incomplete KD, and 2 g/kg IVIG therapy was given. Due to persistent fever, three daily 30 mg/kg per day pulse methylprednisolone and 5 mg/kg per day oral cyclosporine were added to the therapy. Her fever and all clinical symptoms were resolved. Corticosteroid therapy was tapered gradually and ceased at the end of the third month of the treatment. Afterwards, the patient did not come to our clinic for follow-up visits. The patient was referred to our clinic again at the age of 8 with complaints of fever, oral aphthous stomatitis, maculo-papular skin rash, and abdominal pain. We learned that she had tonsillectomy due to the diagnosis of periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) syndrome in another hospital because of recurrent tonsillitis attacks at the age of 4, but her febrile attacks did not regress. Her laboratory, clinical, and bone marrow findings were compatible with MAS again. Pulse methylprednisolone and IVIG treatments were started, and her symptoms was resolved promptly. Based on clinical and laboratory findings including recurrent attacks of MAS preceded by recurrent fever episodes, aphthous stomatitis, tonsillitis, cervical lymphadenopathy, maculo-papular rash, headache, and elevated acute phase reactants since the age of 6-month-old, homozygous V377I mutation was on MVK gene, and a final diagnosis of HIDS was made in this patient. She well responded to anakinra, an IL-1 receptor blocker, and has been followed-up without any complaints associated inflammatory attacks for 3 months.
Case 2
A 27-month-old girl was admitted to the pediatric rheumatology clinic with prolonged fever, skin rash, arthralgia, and myalgia lasting for 15 days. Physical examination showed maculopapular skin rash on trunk, face, and extremities, edema on the extensor surface of the feet and hands, a 1.5 cm diameter of cervical lymphadenopathy, abdominal pain, and hepatosplenomegaly. There was no consanguinity between parents. Leukopenia, anemia, elevated ferritin, alanine aminotransferase and triglyceride levels, and high acute phase reactants were detected. ANA, anti-dsDNA, RF and brucella tests were negative; serum complement levels and viral serology tests were normal. Chest X-ray and echocardiography were normal. The patient was diagnosed as sJIA with the signs of prolonged fever, rash, hepatosplenomegaly, and arthritis. Bone marrow aspiration was compatible with MAS, and a diagnosis of MAS secondary to sJIA was made. Pulse methylprednisolone with a 30 mg/kg per day was prescribed for 3 days, and then 1 mg/kg per day oral prednisolone was continued. Anakinra was added to the treatment due to higher acute phase markers in the third month of the treatment. Her symptoms were resolved, laboratory parameters returned to normal, and corticosteroid therapy was tapered gradually and stopped at the end of 6 months. The patient was admitted to our clinic 3 months later at the age of 36-month-old with complaints of fever, maculopapular skin rash, arthralgia, myalgia, and abdominal pain under anakinra therapy. Her laboratory, clinical, and bone marrow findings were compatible with MAS again. Pulse methylprednisolone and IVIG treatments were started, and her symptoms regressed partially. The patient’s diagnosis was challenged again. An autoinflammatory gene panel was studied, and V377I homozygous mutation was detected on MVK gene, and then the diagnosis of HIDS was made. Canakinumab, which is a monoclonal antibody against IL-1 receptor, was started. The patient has been followed up under canakinumab treatment for 6 months in complete remission.
Both patients’ demographic, clinical, laboratory and treatment features are shown in Table 1. Informed consents were obtained from the parents of the cases.
Search strategy
We searched PubMed using the keywords “recurrent macrophage activation syndrome,” “hyperimmunoglobulin D syndrome,” and “mevalonate kinase deficiency” and reviewed the current literature. We searched the English literature from inception to June 2022. Articles were excluded if patients with HIDS did not experience any MAS attacks. Editorial letters and abstracts were excluded. Patients’ age, gender, clinical findings, MVK gene mutation, treatment strategies, treatment response, clinical findings, and outcomes were recorded for each patient. This paper is prepared as a narrative review.
Discussion
MAS may show a recurrent course with a high mortality rate in many childhood diseases, mainly reported in sJIA, and rarely reported in autoinflammatory diseases [18]. To our knowledge, our cases are the first report in the current literature, presenting with recurrent MAS attacks due to HIDS disease.
Firstly, in 2007, Rigante et al. reported a 7-year-old female patient who was previously diagnosed as HIDS, presenting with persistent fever, serositis, lethargy, heart failure, increased ferritin-triglyceride-interferon γ levels, hypofibrinogenemia, thrombocytopenia, and anemia. Bone marrow aspiration revealed activated macrophages compatible with MAS. A favorable response with complete remission was attained by corticosteroid and CSA treatment [16]. In 2014, Schulert et al. described an 11-month-old patient presenting with non-remitting fever, respiratory distress, abdominal distention, hepatosplenomegaly, hyperbilirubinemia, elevated liver function tests and elevated CRP, thrombocytopenia, and anemia, diagnosed with MKD with a compound heterozygous mutation in MVK gene. Liver biopsy revealed sinusoidal histiocytes and hemophagocytosis, while bone marrow aspiration was unremarkable. The patient well responded to corticosteroid and anakinra therapy [19]. A cohort of 50 MKD patients from France reported that 6 patients developed MAS and one died [20]. In a series of 114 cases with MKD from the Eurofever registry, only one patient had experienced a MAS attack, responded partially to anakinra, while completely to canakinumab treatment [21]. Short-term corticosteroids and NSAIDs are beneficial to relieve the symptoms during inflammatory attacks. Colchicine is ineffective to prevent inflammatory attacks. Anti-IL-1 therapies are the main treatment in HIDS; however, there are some reports about the benefits of anti-TNF therapies. Allogenic hematopoietic stem cell transplantation may be considered in cases unresponsive to current treatments [1]. Our first case achieved a complete remission for inflammatory attacks with anakinra, while the second case with canakinumab, in addition to corticosteroid therapy. A literature review consisting of HIDS cases who complicated with a MAS episode is exhibited in Table 2.
Although the clinical presentation of MAS in the course of childhood rheumatological, infectious, and malignant diseases is relatively rare, it has to be evaluated in the differential diagnosis and appropriate treatment should be promptly started. The presence of recurrent fever attacks, a family history of autoinflammatory disease, parental consanguinity, and a detailed physical examination should carefully be evaluated by clinicians. Thors et al. described an 8-week-old girl patient diagnosed with incomplete KD due to prolonged fever, increased acute phase reactants, and coronary artery aneurysm [22]. At the follow-up, a diagnosis of HIDS was made with the detection of compound heterozygous mutation in MVK gene performed because of recurrent fever attacks. They emphasized that autoinflammatory diseases, like HIDS, should be kept in mind when recurrent fever attacks present in patients initially diagnosed as KD [22]. A survey from France evaluating the initial diagnosis of thirteen patients with MKD reported that a variety of other diseases was suspected before the right diagnosis, such as sJIA, PFAPA, familial Mediterranean fever, chronic infantile neurological cutaneous and articular syndrome, vasculitis, KD, inflammatory bowel disease, and immunodeficiency [23]. According to the new Eurofever/PRINTO classification criteria, a diagnosis of MKD is based on the presence of a bi-allelic MVK gene mutation plus at least one of the followings, gastrointestinal symptoms, cervical lymphadenopathy, and aphthous stomatitis [24]. The diagnosis of sJIA is characterized by prolonged fever lasting at least 2 weeks and arthritis along with generalized lymphadenopathy, serositis, hepatosplenomegaly, and/or maculopapular skin rash [25]. We had initially misdiagnosed our first patient as KD, and then PFAPA, and finally, patient was diagnosed as HIDS due to recurrent fever, aphthous stomatitis, and cervical lymphadenopathy attacks with a documented MVK gene mutation. Our second patient was misdiagnosed as sJIA firstly, but the patient was diagnosed with HIDS after genetic studies due to the recurrence attacks of fever, skin rash, abdominal pain, and arthritis during the follow-up.
The diagnosis of MAS based on Ravelli criteria indicates that sJIA patients with persistent fever and increased ferritin level (> 684 ng/ml), and requirement of 2 criteria of the followings; decreased thrombocyte count (≤ 181 × 109/L), increased AST level (> 48 U/L), decreased fibrinogen level (< 360 mg/dl), and increased triglyceride level (> 156 mg/dl). Application of bone marrow aspiration to demonstrate hemophagocytic cells may be needed in only suspicious patients [26]. Both our patients met the Ravelli criteria. MAS can be fatal in patients with delayed diagnosis and prompt treatment of MAS is critical. Differentiation of MAS from primary hemophagocytic lympho-histiocytosis (HLH), an autosomal recessive inherited disease leading to deficiency of proteins in perforin-dependent pathway of cytolysis, is important because of the differences in the treatment protocols [26]. MH score has been generated for differentiating primary HLH from MAS, comprising six following items, including age at disease onset, neutrophil and platelet counts, fibrinogen level, hemoglobin level, and splenomegaly. A MH score of ≥ 60 is an indicator of primary HLH than MAS with a best performance [27]. MH score of our two patients were lower than 60 and we did not perform a genetic analysis for primary HLH to the patients.
MAS is a hyperinflammatory condition by a cytokine storm, including IL-1, IL-6, IL-18, and TNFα [9]. Therefore, our main aim in treatment of MAS is to suppress this hyperinflammatory condition with immunosuppressive therapy. High-dose corticosteroid and CSA therapy are the most common used agents in the treatment of MAS. Administration of high doses of anakinra which blocks IL-1α and IL-1β and IVIG treatments was reported to be successful, but there is still a lack of controlled studies [17, 26]. We have well controlled our first patient’s recurrent MAS attacks and primary disease activity with anakinra in addition to corticosteroid, CSA, and IVIG treatments. Our second patient well responded to canakinumab in addition to corticosteroid and IVIG treatments. This patient was unresponsive to anakinra.
In conclusion, HIDS should be considered in differential diagnosis in pediatric patients presenting with recurrent MAS attacks. In these patients, clinicians should be vigilant in terms of the underlying autoinflammatory disease phenotype at the follow-up visits, and corticosteroids with anti-IL-1 treatments are beneficial treatment options.
Data availability
Data sharing is not applicable — no new data is generated, or the article describes entirely case report.
References
Zhang S (2016) Natural history of mevalonate kinase deficiency: a literature review. Pediatr Rheumatol Online J 14:30. https://doi.org/10.1186/s12969-016-0091-7
Drenth JP, Cuisset L, Grateau G, Vasseur C, van de Velde-Visser SD, de Jong JG, Beckman JS, van der Meer JW, Delpech M (1999) Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. Nat Genet 22:178–181. https://doi.org/10.1038/9696
Rigante D, Frediani B, Cantarini L (2018) A comprehensive overview of the hereditary periodic fever syndromes. Clin Rev Allergy Immunol 54:446–453. https://doi.org/10.1007/s12016-016-8537-8
van der Hilst JC, Bodar EJ, Barron KS, Frenkel J, Drenth JP, van der Meer JW, Simon A, International HIDS Study Group (2008) Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine (Baltimore) 87:301–310. https://doi.org/10.1097/MD.0b013e318190cfb7
ter Haar NM, Oswald M, Jeyaratnam J, Anton J, Barron KS, Brogan PA et al (2015) Recommendations for the management of autoinflammatory diseases. Ann Rheum Dis 74:1636–1644. https://doi.org/10.1136/annrheumdis-2015-207546
Rigante D, Ansuini V, Bertoni B, Pugliese AL, Avallone L, Federico G, Stabile A (2006) Treatment with anakinra in the hyperimmunoglobulinemia D/periodic fever syndrome. Rheumatol Int 27:97–100. https://doi.org/10.1007/s00296-006-0164-x
Takada K, Aksentijevich I, Mahadevan V, Dean JA, Kelley RI, Kastner DL (2003) Favorable preliminary experience with etanercept in two patients with the hyperimmunoglobulinemia D and periodic fever syndrome. Arthritis Rheum 48:2645–2651. https://doi.org/10.1002/art.11218
Malcova H, Strizova Z, Milota T, Striz I, Sediva A, Cebecauerova D, Horvath R (2021) IL-1 Inhibitors in the treatment of monogenic periodic fever syndromes: from the past to the future perspectives. Front Immunol 11:619257. https://doi.org/10.3389/fimmu.2020.619257
Brisse E, Matthys P, Wouters CH (2016) Understanding the spectrum of haemophagocytic lymphohistiocytosis: update on diagnostic challenges and therapeutic options. Br J Haematol 174:175–187. https://doi.org/10.1111/bjh.14144
Aytac S, Batu ED, Unal S et al (2016) Macrophage activation syndrome in children with systemic juvenile idiopathic arthritis and systemic lupus erythematosus. Rheumatol Int 36:1421–1429. https://doi.org/10.1007/s00296-016-3545-9
Barut K, Adrovic A, Sahin S, Tarcin G, Tahaoglu G, Koker O, Yildiz M (2019) Kasapcopur O (2019) Prognosis, complications and treatment response in systemic juvenile idiopathic arthritis patients: a single-center experience. Int J Rheum Dis 22:1661–1669. https://doi.org/10.1111/1756-185X.13649
Bossi G, Codazzi AC, Vinci F, Clerici E, Regalbuto C, Crapanzano C, Veraldi D, Moiraghi A, Marseglia GL (2022) Efficacy of anakinra on multiple coronary arteries aneurysms in an infant with recurrent Kawasaki disease, complicated by macrophage activation syndrome. Children (Basel) 9:672. https://doi.org/10.3390/children9050672
Horneff G, Rhouma A, Weber C, Lohse P (2013) Macrophage activation syndrome as the initial manifestation of tumour necrosis factor receptor 1-associated periodic syndrome (TRAPS). Clin Exp Rheumatol 31(3 Suppl 77):99–102
Çağlayan Ş, Ulu K, Çakan M, Sözeri B (2022) A rare onset in tumor necrosis factor receptor-associated periodic syndrome: recurrent macrophage activation syndrome triggered by COVID-19 infection. Rheumatology (Oxford). Jun 22:keac362. https://doi.org/10.1093/rheumatology/keac362
Iyengar VV, Chougule A, Gowri V, Taur P, Prabhu S, Bodhanwala M, Desai MM (2022) DADA2 presenting as nonimmune hemolytic anemia with recurrent macrophage activation syndrome. Pediatr Blood Cancer 69:e29461. https://doi.org/10.1002/pbc.29461
Rigante D, Capoluongo E, Bertoni B, Ansuini V, Chiaretti A, Piastra M, Pulitanò S, Genovese O, Compagnone A, Stabile A (2007) First report of macrophage activation syndrome in hyperimmunoglobulinemia D with periodic fever syndrome. Arthritis Rheum 56:658–661. https://doi.org/10.1002/art.22409
Sönmez HE, Demir S, Bilginer Y, Özen S (2018) Anakinra treatment in macrophage activation syndrome: a single center experience and systemic review of literature. Clin Rheumatol 37:3329–3335. https://doi.org/10.1007/s10067-018-4095-1
Stabile A, Bertoni B, Ansuini V, La Torraca I, Sallì A, Rigante D (2006) The clinical spectrum and treatment options of macrophage activation syndrome in the pediatric age. Eur Rev Med Pharmacol Sci 10:53–59
Schulert GS, Bove K, McMasters R, Campbell K, Leslie N, Grom AA (2014) Mevalonate kinase deficiency associated with recurrent liver dysfunction, macrophage activation syndrome and perforin gene polymorphism. Arthritis Care Res (Hoboken) 67:1173–1179. https://doi.org/10.1002/acr.22527
Bader-Meunier B, Florkin B, Sibilia J et al (2011) Mevalonate kinase deficiency: a survey of 50 patients. Pediatrics 128:152–159. https://doi.org/10.1542/peds.2010-3639
Ter Haar NM, Jeyaratnam J, Lachmann HJ et al (2016) The phenotype and genotype of mevalonate kinase deficiency: a series of 114 cases from the Eurofever registry. Arthritis Rheumatol 68:2795–2805. https://doi.org/10.1002/art.39763
Thors VS, Vastert SJ, Wulffraat N, van Royen A, Frenkel J, Velden M, de Sain-van der, de Koning TJ (2014) Periodic fever in MVK deficiency: a patient initially diagnosed with incomplete Kawasaki disease. Pediatrics 133:461–465. https://doi.org/10.1542/peds.2012-1372
Berody S, Galeotti C, Koné-Paut I, Piram M (2015) A restrospective survey of patients’s journey before the diagnosis of mevalonate kinase deficiency. Joint Bone Spine 82:240–244. https://doi.org/10.1016/j.jbspin.2014.12.011
Gattorno M, Hofer M, Federici S et al Eurofever Registry and the Paediatric Rheumatology International Trials Organisation (PRINTO) (2019) Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis 78:1025–1032. https://doi.org/10.1136/annrheumdis-2019-215048
Lee JJY, Schneider R (2018) Systemic juvenile idiopathic arthritis. Pediatr Clin North Am 65:691–709. https://doi.org/10.1016/j.pcl.2018.04.005
Ravelli A, Davì S, Minoia F, Martini A, Cron RQ (2015) Macrophage activation syndrome. Hematol Oncol Clin North Am 29:927–941. https://doi.org/10.1016/j.hoc.2015.06.010
Minoia F, Bovis F, Davì S et al (2017) Development and initial validation of the macrophage activation syndrome/primary hemophagocytic lymphohistiocytosis score, a diagnostic tool that differentiates primary hemophagocytic lymphohistiocytosis from macrophage activation syndrome. J Pediatr 189:72–78. https://doi.org/10.1016/j.jpeds.2017.06.005
Acknowledgements
The authors are grateful to the patient and his family.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosures
None.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gezgin Yıldırım, D., Yıldız Yıldırım, Ç., Karaçayır, N. et al. Recurrent macrophage activation syndrome due to hyperimmunoglobulin D syndrome: a case-based review. Clin Rheumatol 42, 277–283 (2023). https://doi.org/10.1007/s10067-022-06384-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-022-06384-9