
Abstract
This study aimed to examine whether smoking behavior is causally related to gout. Summary statistics of publicly available data from genome-wide association studies (GWAS) of smoking behavior (n = 85,997) served as the exposure dataset, while meta-analysis results of 14 studies including 2115 cases and 67,259 controls of European descent served as the outcome dataset. The data were subjected to two-sample Mendelian randomization (MR) analysis using the inverse-variance weighted (IVW), weighted median, and MR-Egger regression methods. Five single-nucleotide polymorphisms (SNPs) from GWAS of smoking behavior were selected as instrumental variables (IVs) to improve inference: CHRNA3 (rs1051730), PDE1C (rs215614), CYP2A6 (rs4105144), CHRNB3 (rs6474412), and CYP2B6 (rs7260329). The IVW data did not support a causal association between smoking behavior and gout (beta = − 0.035, SE = 0.036, p = 0.333). MR-Egger regression indicated that directional pleiotropy did not bias the result (intercept = 0.021; p = 0.560). MR-Egger analysis revealed no causal association between smoking behavior and gout (beta = − 0.074, SE = 0.070, p = 0.366). The weighted median approach did not support a causal association between smoking behavior and gout (beta = − 0.043, SE = 0.040, p = 0.279). Cochran’s Q test indicated no evidence of heterogeneity between IV estimates based on individual variants. The results of “leave one out” analysis demonstrated that no single SNP drove the IVW point estimate. MR estimates using IVW, weighted median, and MR-Egger analysis were consistent and did not support a causal inverse association between smoking behavior and gout.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Gout is an inflammatory disorder characterized by hyperuricemia and deposition of monosodium urate crystals in intra-articular and peri-articular locations, resulting in episodic gout flares, gouty arthropathy, tophi formation, and urolithiasis [1]. Although the etiology of gout is not fully understood, it is clear that gout has both genetic and environmental components [2, 3]. The primary cause of gout is hyperuricemia due to excess urate production or impaired renal excretion of uric acid. Increased levels of hyperuricemia are correlated with a higher incidence of gout [4].
Many environmental factors have been suggested to induce gout [2]. Hyperuricemia can be influenced by risk factors such as age, obesity, sex, hypertension, kidney disease, alcohol intake, and cigarette smoking [2]. Among these factors, smoking has been reported to have a potentially protective association with gout, although the evidence is inconsistent [5,6,7]. Smoking is considered to impact the immune system and may have both pro-inflammatory and anti-inflammatory effects [8]. Smoking is associated with lower serum urate concentrations, possibly by inactivating xanthine oxidase via the cyanides in cigarettes [9]. However, observational studies are prone to bias such as reverse causation interpretations and residual confounding, preventing a clear understanding of the effect of smoking on gout [10, 11]. Cigarette smoking is correlated with lower body mass, alcohol intake, and higher prevalence of hypertension, dyslipidemia, and kidney disease, which can influence the risk of gout [12,13,14].
Mendelian randomization (MR) is a technique that uses genetic variants as instrumental variables (IVs) to assess whether an observational association between a risk factor and outcome is consistent with a causal effect [15]. MR analysis can be used to evaluate whether genetically increased smoking behavior is associated with a decreased risk of gout. Two-sample MR estimates causal effects between datasets for the exposure and outcome variable measured in independent samples, which is a very useful method for situations in which it is difficult to measure the exposure and outcome in the same set of individuals [16]. However, MR has not been applied to explore causal effects of smoking on gout risk. Therefore, the aim of the present study was to examine whether smoking behavior is causally associated with gout using two-sample MR analysis.
Materials and methods
Data sources and selection of genetic variants
We searched the NHGRI-EBI GWAS catalog (https://www.ebi.ac.uk/gwas/), which is a comprehensive catalog of reported associations from published genome-wide association studies (GWAS). As the exposure dataset, we used publicly available summary statistics (total n = 85,997) of GWAS meta-analyses to determine the association of the number of cigarettes smoked per day (CPD) in smokers (n = 31,266) and smoking initiation (n = 46,481) using samples from the ENGAGE Consortium, with replication in the Tobacco and Genetics and Glaxo Smith Kline consortia cohorts (n = 45,691 smokers), and a third sample of European ancestry (n = 9040) [17]. The outcome dataset included summary statistics datasets of the meta-analysis of gout comprising 14 studies including 2115 cases and 67,259 controls of European ancestry [18]. A two-sample MR study using genetic variants associated with gout as IVs was also performed to improve inference. We obtained summary statistics (beta coefficients and standard errors) for five single-nucleotide polymorphisms (SNPs) associated with smoking behavior as IVs from smoking behavior GWAS [17]. Additionally, we utilized summary data for five SNPs from gout GWAS as the outcome dataset [19].
Statistical analysis for MR
MR analysis requires genetic variants that may be related to, but are not potential confounders of, an exposure variable [20]. First, we assessed the independent association of four SNPs with smoking behavior. Second, we examined the association between each SNP and the risk of gout. Third, we combined these findings to estimate the uncompounded causal association between smoking behavior and gout risk by MR analysis. As described above, we performed two-sample MR to estimate the causal effect of exposure (smoking) on the outcome (gout) using summary statistics from different GWAS [21] to assess the causal relationships between smoking behavior and gout risk, using summary data from smoking behavior and gout GWAS with five SNPs as IVs.
The inverse variance-weighted (IVW) method uses a meta-analysis approach to combine Wald ratio estimates of the causal effect obtained from associations of different SNPs and provides a consistent estimate of the causal effect of exposure on the outcome when each genetic variant satisfies the assumptions of an IV [22]. Although including multiple variants in MR analysis results in increased statistical power, pleiotropic genetic variants that are not valid IVs may also be included [21]. To explore and adjust for such pleiotropy effects (i.e., association of genetic variants with more than one variable), weighted median and MR-Egger regression methods were performed. MR-Egger regression analysis tests and accounts for the presence of unbalanced pleiotropy by introducing a parameter for this bias by incorporating summary data estimates of the causal effects with multiple individual variants, and has been shown to be robust against invalid instruments [23]. The MR-Egger procedure performs weighted linear regression of the gene-outcome coefficients on gene-exposure coefficients [23]. The slope of this regression represents the causal effect estimate, and the intercept can be interpreted as an estimate of the average horizontal pleiotropic effect across all genetic variants [24]. The weighted median estimator then provides a consistent estimate of the causal effect, even when up to 50% of the information contributing to the analysis is derived from genetic variants that are invalid IVs [25]. Use of the weighted median estimator increases the precision of the estimates compared to MR-Egger analysis [25]. All tests were considered statistically significant at p < 0.05. All MR analyses were performed using the MR-Base platform [26].
Heterogeneity and sensitivity testing
We assessed heterogeneities among SNPs using Cochran’s Q-statistics and funnel plots [27]. We also performed a “leave one out” analysis to investigate whether the causal association observed was driven by a unique SNP. Additionally, subgroup analysis was performed using only IV SNPs at a genome-wide significance level for the sensitivity test.
Results
Studies included in the meta-analysis
Instrumental variables for MR
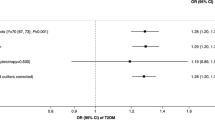
We selected five SNPs from smoking behavior GWAS as IVs [17]. These SNPs were in the genes CHRNA3 (rs1051730), PDE1C (rs215614), CYP2A6 (rs4105144), CHRNB3 (rs6474412), and CYP2B6 (rs7260329) (Table 1, Fig. 1). Except for rs215614 and rs7260329, the SNPs were found to be associated with smoking behavior at a genome-wide significance level and were inversely associated with gout. However, none of these SNPs were nominally significantly associated with gout (Table 1).

Forest plot of the causal effects of smoking behavior-associated single-nucleotide polymorphisms on gout
Mendelian randomization results
The IVW method showed no evidence to support a causal association between smoking behavior and gout (beta = − 0.035, SE = 0.036, p = 0.333) (Table 2, Figs. 1 and 2). The intercept represents the average pleiotropic effect across the genetic variants (average direct effect of a variant with the outcome). An intercept differing from zero (MR-Egger test) is evidence of directional pleiotropy. MR-Egger regression revealed that directional pleiotropy was unlikely to have biased the result (intercept = 0.021; p = 0.560). MR-Egger analysis revealed no causal association between smoking behavior and gout (beta = − 0.074, SE = 0.070, p = 0.366) (Table 2, Figs. 1 and 2). The weighted median approach yielded no evidence of a causal association between smoking behavior and gout (beta = − 0.043, SE = 0.040, p = 0.279) (Table 2, Fig. 2). The MR estimates determined using the IVW, weighted median, and MR-Egger regression methods were consistent. Therefore, the MR analysis results did not support a causal inverse association between smoking behavior and gout.

Scatter plots of genetic associations with smoking behavior against the genetic associations with gout (gout). The slopes of each line represent the causal association for each method. The blue line represents the inverse variance-weighted (IVW) estimate, the green line represents the weighted median estimate, and the dark blue line represents the MR-Egger estimate
Heterogeneity and sensitivity tests
Cochran’s Q test indicated no evidence of heterogeneity among the IV estimates based on the individual variants (Table 2). The results of “leave one out” analysis demonstrated that no single SNP was driving the IVW point estimate for gout (Fig. 3). Additionally, when the MR analysis was limited to only three SNPs (rs1051730, rs4105144, and rs6474412) at the genome-wide significance level, the results remained non-significant (IVW beta = − 0.048, SE = 0.038, p = 0.207).

“Leave one out” analysis to investigate whether the causal association was driven by a unique single-nucleotide polymorphism (SNP)
Discussion
Epidemiologic studies suggested that smoking is associated with a lower risk of gout in men, although there are also conflicting findings [6, 7]. It is possible that smoking impacts serum urate and gout risk via mechanisms in which cigarette smoking causes oxidative stress, leading to the depletion of uric acid, which is one of the most important antioxidant scavengers of reactive oxygen species [28], and/or that cyanide in smoking may inhibit xanthine oxidase for urate formation in purine metabolism [9]. The causal relationship between smoking and gout remains unclear because previously reported associations may have been influenced by biases or confounding factors inherent to observational studies, such as reverse causation interpretation, a low number of studies of small sizes, and selection bias [10, 29]. Thus, to clarify this association, we evaluated causality using three different estimation methods (IVW method, weighted median method, and MR-Egger regression) for MR analyses. Our study did not indicate that the inverse associations between gout and Alzheimer’s disease are causal. The MR estimates using IVW, MR Egger, and weighted median analysis were consistent and do not support a causal inverse association between smoking behavior and gout.
MR minimizes the possibility of the bias inherent to observational studies due to residual confounding or reverse causality [30]. However, MR studies are susceptible to bias from pleiotropy (association of genetic variants with more than one variable) [31]. Although including multiple variants in MR analysis typically leads to increased statistical power, it also may include pleiotropic genetic variants that are not valid IVs [32]. Therefore, appropriate sensitivity analysis is essential for testing the validity of any conclusions drawn from an MR study. Here, to eliminate the influence of pleiotropy, we employed a weighted median estimator, which provides valid estimates even if 50% of the SNPs are not valid instruments [25]. We also used MR-Egger regression to test for unbalanced pleiotropy and determine a causal estimate of the influence of exposure on the outcome [23]. Our results using all three approaches were consistent, and the MR-Egger approach showed no evidence of unbalanced pleiotropy as indicated by the intercept p value. The MR-Egger method results in a loss of precision and power, but our weighted median estimator results were similar to the IVW estimator, providing additional confidence in these findings.
The present study has several limitations. First, our analysis included a relatively small number of SNPs as IVs and thus may have had limited power in detecting an association. The statistical power can be increased and a more precise causal estimate can be obtained by combining multiple genetic variants [33]. Second, genetic variants have only a modest effect on a given exposure (smoking behavior) because they may only explain a very small proportion of the overall variance in a particular exposure [34]. Thus, very large numbers of cases are required to accurately detect a causal relationship for the outcome of interest. Third, it is important to ensure that there is a strong relationship between the genetic variant and an exposure. “Weak instrument bias,” which refers to a genetic variant that does not have a sufficiently strong association with the exposure, may affect MR analysis [35]. However, the sensitivity test performed to limit our analysis to SNPs at a genome-wide significance level did not change the results. Fourth, the gout dataset was based on a study that included participants of European ancestry. As causality may depend on ethnicity and selection bias, further MR studies of other populations are needed. Fifth, epidemiologic data showed that cigarette smoking is associated with a decreased risk of gout, particularly among men [6, 7]. The association between cigarette smoking and gout is not discernible among women. We could not analyze our MR based on gender. Nevertheless, this meta-analysis also has strengths. Although smoking has been investigated as a potential risk factor for gout, this is the first MR to assess this association. It is important that estimates of both gene-exposure and gene-outcome associations are available for each variant considered. Thus, this is the first study to determine the causal relationship between smoking behavior and gout.
In conclusion, the MR analysis results do not support inverse causal associations between smoking behavior and gout development. Epidemiological evidence for a relationship between smoking behavior and decreased risk of gout does not appear to be causal. However, we cannot rule out small associations. Well-designed epidemiological studies and MR studies using more variants that explain a greater proportion of smoking behaviors are warranted to confirm or rule out the causal relationship.
References
Wortmann RL (2002) Gout and hyperuricemia. Curr Opin Rheumatol 14:281–286
Doherty M (2009) New insights into the epidemiology of gout. Rheumatology 48:ii2–ii8
Lee YH, Song GG (2012) Pathway analysis of genome-wide association studies on uric acid concentrations. Hum Immunol 73:805–810
Terkeltaub RA (2003) Gout. N Engl J Med 349:1647–1655
Fanning N, Merriman TR, Dalbeth N, Stamp LK (2017) An association of smoking with serum urate and gout: a health paradox. Semin Arthritis Rheum 47:825–842
Gee Teng G, Pan A, Yuan JM, Koh WP (2016) Cigarette smoking and the risk of incident gout in a prospective cohort study. Arthritis Care Res 68:1135–1142
Wang W, Krishnan E (2014) Cigarette smoking is associated with a reduction in the risk of incident gout: results from the Framingham Heart Study original cohort. Rheumatology 54:91–95
Arnson Y, Shoenfeld Y, Amital H (2010) Effects of tobacco smoke on immunity, inflammation and autoimmunity. J Autoimmun 34:J258–J265
Mouhamed DH, Ezzaher A, Neffati F, Douki W, Gaha L, Najjar MF (2011) Effect of cigarette smoking on plasma uric acid concentrations. Environ Health Prev Med 16:307–312
Jepsen P, Johnsen S, Gillman M et al (2004) Interpretation of observational studies. Heart 90:956–960
Song GG, Bae S-C, Lee YH (2012) Association between vitamin D intake and the risk of rheumatoid arthritis: a meta-analysis. Clin Rheumatol 31:1733–1739
Virdis A, Giannarelli C, Fritsch Neves M, Taddei S, Ghiadoni L (2010) Cigarette smoking and hypertension. Curr Pharm Des 16:2518–2525
Júnior E, Fernando U, Elihimas HCS, Lemos VM, Leão MA, Sá MPBO, França EETD, Lemos A, Valente LM, Markman Filho B (2014) Smoking as risk factor for chronic kidney disease: systematic review. J Bras Nefrol 36:519–528
Kim S-K (2018) Interrelationship of uric acid, gout, and metabolic syndrome: focus on hypertension, cardiovascular disease, and insulin resistance. J Rheum Dis 25:19–27
Burgess S, Daniel RM, Butterworth AS, Thompson SG, Consortium E-I (2014) Network Mendelian randomization: using genetic variants as instrumental variables to investigate mediation in causal pathways. Int J Epidemiol 44:484–495
Lawlor DA (2016) Commentary: two-sample Mendelian randomization: opportunities and challenges. Int J Epidemiol 45:908–915
Thorgeirsson TE, Gudbjartsson DF, Surakka I, Vink JM, Amin N, Geller F, Sulem P, Rafnar T, Esko T, Walter S (2010) Sequence variants at CHRNB3–CHRNA6 and CYP2A6 affect smoking behavior. Nat Genet 42:448–453
Köttgen A, Albrecht E, Teumer A, Vitart V, Krumsiek J, Hundertmark C, Pistis G, Ruggiero D, O'Seaghdha CM, Haller T (2013) Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat Genet 45:145–154
Consortium a (2012) Identification of new susceptibility loci for osteoarthritis (arcOGEN): a genome-wide association study. Lancet 380:815–823
Burgess S, Butterworth A, Thompson SG (2013) Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol 37:658–665
Hartwig FP, Davies NM, Hemani G, Davey Smith G (2016) Two-sample Mendelian randomization: avoiding the downsides of a powerful, widely applicable but potentially fallible technique. Int J Epidemiol 45:1717–1726
Pierce BL, Burgess S (2013) Efficient design for Mendelian randomization studies: subsample and 2-sample instrumental variable estimators. Am J Epidemiol 178:1177–1184
Bowden J, Davey Smith G, Burgess S (2015) Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 44:512–525
Burgess S, Thompson SG (2017) Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol 32:377–389
Bowden J, Davey Smith G, Haycock PC, Burgess S (2016) Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 40:304–314
Hemani G, Zheng J, Wade K, Elsworth B, Langdon R, Burgess S (2016) MR-Base: a platform for systematic causal inference across the phenome using billions of genetic associations. bioRxiv 16:78972
Egger M, Smith GD, Phillips AN (1997) Meta-analysis: principles and procedures. BMJ 315:1533–1537
Tsuchiya M, Asada A, Kasahara E, Sato EF, Shindo M, Inoue M (2002) Smoking a single cigarette rapidly reduces combined concentrations of nitrate and nitrite and concentrations of antioxidants in plasma. Circulation 105:1155–1157
Lee YH, Bae SC, Song GG (2013) Hepatitis B virus (HBV) reactivation in rheumatic patients with hepatitis core antigen (HBV occult carriers) undergoing anti-tumor necrosis factor therapy. Clin Exp Rheumatol 31:118–121
Smith DG (2011) Mendelian randomization for strengthening causal inference in observational studies: application to gene X environment interactions. Psychol Sci 6:314–314
Thompson JR, Minelli C, Bowden J, Del Greco FM, Gill D, Jones EM, Shapland CY, Sheehan NA (2017) Mendelian randomization incorporating uncertainty about pleiotropy. Stat Med 36:4627–4645
Smith GD, Ebrahim S (2004) Mendelian randomization: prospects, potentials, and limitations. Int J Epidemiol 33:30–42
Burgess S, Dudbridge F, Thompson SG (2016) Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat Med 35:1880–1906
Swerdlow DI, Kuchenbaecker KB, Shah S, Sofat R, Holmes MV, White J, Mindell JS, Kivimaki M, Brunner EJ, Whittaker JC (2016) Selecting instruments for Mendelian randomization in the wake of genome-wide association studies. Int J Epidemiol 45:1600–1616
Burgess S, Thompson SG, Collaboration CCG (2011) Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol 40:755–764
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosures
None.
Rights and permissions
About this article

Cite this article
Lee, Y.H. Assessing the causal association between smoking behavior and risk of gout using a Mendelian randomization study. Clin Rheumatol 37, 3099–3105 (2018). https://doi.org/10.1007/s10067-018-4210-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-018-4210-3