
Abstract
Thrombotic thrombocytopenic purpura (TTP) is a potentially lethal multisystem disorder which could be caused by autoimmune diseases. However, the concomitant occurrence of TTP and Sjögren’s syndrome (SS) is an extremely uncommon scenario, especially in male patients. A 56-year-old Chinese male was admitted for the appearance of diffuse ecchymosis. Then he gradually developed transient slurred speech, progressive confusion, agitation, extremity weakness, and fever. Laboratory investigations suggested anemia, thrombocytopenia, significantly increased lactic dehydrogenase, schistocytes in peripheral blood smear, and a disintegrin-like metalloproteinase with thrombospondin motif type 1 member 13 (ADAMTS13) activity deficiency with high inhibitor titers. TTP was thus diagnosed. The patient also had positive anti-nuclear antibody, anti-SSA, and anti-SSB; however, anti-double stranded DNA (dsDNA) was negative. These drove us to perform ocular and dental sicca evaluation and the finial diagnosis was TTP secondary to SS. Plasma exchange and corticosteroid therapy were effective to control TTP. Cyclophosphamide was subsequently added when the platelet count was stable. The total duration of corticosteroid and cyclophosphamide was 8 and 6 months, respectively. The patient recovered without relapse at 1-year follow-up. To our knowledge, this was the first case of SS initially presented as TTP in a male patient. The case also elucidated the importance of autoantibody screen in the workup of TTP and the benefits of adjunctive immunosuppressive therapy in relapse prevention.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Thrombotic thrombocytopenic purpura (TTP) is a potentially lethal multisystem disorder, with an incidence of approximately five per million per year [17]. As a distinct entity of thrombotic microangiopathy, TTP is characterized by a pentad of clinical presentations, including microangiopathic hemolytic anemia, thrombocytopenia, neurological symptoms, renal impairment, and fever [29]. Sometimes, the manifestations can be nonspecific and the affected organs can be variable [17]. Laboratory assays that support the diagnosis are the presence of schistocytes in peripheral blood smear, the rapid drop of platelet count (PLT), the markedly increase of lactic dehydrogenase (LDH), normal coagulation test, and negative Coombs test [29]. Accumulating evidence have proved that TTP is attributed to a disintegrin-like metalloproteinase with thrombospondin motif type 1 member 13 (ADAMTS13) activity deficiency, which may be either congenital or acquired. Reduced ADAMTS-13 activity impedes the physiological degradation of large von Willebrand factor-(vWF) multimers, further leading to the aggregation of platelet and the widespread microthrombi [35]. This process can be provoked by a series of infection, malignancy, autoimmune disease, transplantation, pregnancy, and drugs [39]. Sjögren’s syndrome (SS) is rare on this list, and TTP secondary to SS in male patients is exceedingly rare. Herein we describe a case of a male who suffered from SS initially presenting as TTP and recovered without relapse under the treatment of plasma exchange, corticosteroid pulse, and cyclophosphamide.
Case report
A 56-year-old Chinese male presented with a 1-week history of the appearance of diffuse ecchymosis over the limbs and upper chest. Four days later, he complained of transient slurred speech without headache or seizure. Then he gradually developed progressive confusion, agitation, and extremity weakness (muscle strength 3/6), accompanied by a fever of 38.2 °C. The patient had a 2-year previous history of coronary heart disease with aspirin 0.1 g once daily. Physical examination was otherwise normal.
On admission, laboratory investigations indicated the following: hemoglobin (HGB) 6.2 g/dl, PLT 7 × 108/dl, reticulocyte 156.10 × 108/dl (7.12%), leukocyte count 6.26 × 108/dl, prothrombin time (PT) 12.9 s, activated partial thromboplastin time 36.8 s, serum total bilirubin 1.97 mg/dl (indirect 0.44 mg/dl), LDH 140.2 U/dl, C reactive protein 2.49 mg/dl, erythrocyte sedimentation rate 108 mm/h, blood urea nitrogen 19.00 mg/dl, creatinine 1.16 mg/dl, urobilinogen 0.95 mg/dl, and urine blood trace. Peripheral blood smear was notable for anisocytosis and poikilocytosis with numerous schistocyte, spherocyte, and polychromatic erythrocytes. Bone marrow showed active proliferation with normal granulocytes/nucleated erythrocytes ratio. ADAMTS13 activity was 0% with the presence of high titers of inhibitor. Coombs’ test, Ham’s test, serum, and urine protein electrophoresis were all negative. Antibody screening was performed and the results were as follows: anti-nuclear antibody (ANA) speckle pattern 1:320 (indirect immunofluorescence method, normal range < 1:40), anti-SSA 1:4, and anti-SSB 1:4 (counterimmunoelectrophoresis method, normal range: negative), while anti-Smith, anti-double-strand DNA (dsDNA), lupus anticoagulant, and anti-neutrophil cytoplasmic antibody were all negative. Other immunological studies were as follows: immunoglobulin G 1885 mg/dl (normal range: 700–1700 mg/dl), complement 3 55.8 mg/dl (normal range: 73–146 mg/dl), and complement 4 8.6 mg/dl (normal range: 10–40 mg/dl). Blood cultures, serology (hepatitis B virus, hepatitis C virus, cytomegalovirus, Epstein-Barr virus), and cerebrospinal fluid test did not reveal any signs of infection. Cranial magnetic resonance imaging was unremarkable for hemorrhage or infarction.
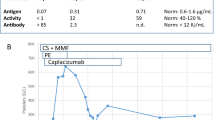
A diagnosis of acquired TTP was confirmed based on the combination of microangiopathic hemolytic anemia, thrombocytopenia, acute neurological abnormalities, fever, and a severe deficiency of ADAMTS13 activity as well as the presence of inhibitors. Before the diagnosis of TTP, the patient was initially treated with intravenous immunoglobulin (IVIG) 20 g daily for 4 consecutive days, whereas the PLT, HGB, and LDH remained unresponsive (Fig. 1). On hospital day 5, when the diagnosis was definite, daily plasma exchange therapy (PET) was initiated in conjunction with steroids (intravenous methylprednisolone 40 mg twice a day), which was switched to prednisone (60 mg daily per oral) 5 days later. Consciousness level was significantly ameliorated and PLT began to increase. After 10-day PET, PLT was elevated to 117 × 108/dl. HGB and LDH returned to normal range (Fig. 1) and no schistocyte was found in peripheral blood smear.

The change of HGB, PLT, LDH, and treatment interventions during hospital course. HGB hemoglobin, PLT platelet, LDH lactic dehydrogenase, IVIG intravenous immunoglobulin, PET plasma exchange treatment, CTX cyclosporine
The positive anti-SSA and anti-SSB antibodies leaded to the suspicion of SS. When further questioned about the history, the patient reported having a recurrent sensation of sand in eyes, yet he paid little attention. Ophthalmology examinations showed positive Schirmer’s test (right 4 mm, left 7 mm), positive fluorescein corneal staining (right 4′, left 5′) and shortened tear break-up time (right 8 s/5 min, left 5 s/5 min). The unstimulated whole salivary flow rate was 0.3 mL/min. Parotid gland radiography revealed very low uptake of the contrast agent in end ducts. Salivary gland biopsy indicated lymphocytic aggregates (focus score 1/4 mm2) and fibrous tissue proliferation. According to the 2016 American College of Rheumatology/European League Against Rheumatism classification criteria [36], a diagnosis of primary SS was confirmed. A series of examinations were carried out to evaluate possible extra-glandular involvement. Thoracic and abdominal radiology, liver function, thyroid function, and renal tubule function tests were all normal. Intravenous cyclophosphamide (CTX) 0.4 g weekly was administrated on hospital day 16 when PLT and HGB were stable.
The patient was discharged on hospital day 23 (with a cumulative iv CTX dose of 0.8 g). Afterwards, CTX was given 0.1 g every other day per oral, considering the inconvenience of intravenous injection outside hospital [3, 6, 14, 15, 31]. When a 4-week full-dose (60 mg/day) prednisone regimen was completed, withdrawal was started in decrements of 5 mg every week until the dose reached 30 mg/day. Then prednisone dose was tapered by 2.5 mg every week at dose 15–30 mg/day, followed by 2.5 mg every 2 weeks until the discontinuation of steroids (the total duration of steroid therapy was approximately 8 months). CTX was used for 6 months and was discontinued because laboratory tests showed stable level of HGB and PLT. The patient was asymptomatic after discharge. Blood tests were taken every 3 months which revealed normal hemoglobin, platelet, and LDH level, as well as normal ADAMTS13 activity. During the 1-year follow-up, no signs of TTP relapse were observed and the patient was free of medication and enjoyed his previous normal lifestyle.
Discussion and literature review
The spectrum of autoimmune diseases that may cause acquired TTP is wide, including systemic lupus erythematosus, anti-phospholipid antibody syndrome, mixed connective tissue disease, rheumatoid arthritis, scleroderma, and dermatomyositis [2]. However, the concomitant occurrence of TTP and SS is an extremely uncommon scenario. To the best of our knowledge, only 11 other cases of SS complicated with TTP have been reported in nine English articles in the recent 40 years (Table 1) [1, 9, 18, 20, 23, 32, 37, 38, 41]. Although the present case shares similarities with those ones, we still believed that it uniquely illustrates this rare medical condition from the following aspects.
First, this is the first case of SS-related TTP in male patients. Given its autoimmune nature, both TTP and SS have a female to male predominance (2:1–3:2 in TTP and 9:1–16:1 in SS) [7, 12, 22]. Actually, SS might be caused partly by differential function of sex hormone on immune cells [7]. It has been hypothesized that the lack of androgens effect in the exocrine glands may trigger acinar cell damage [26, 27]. The atypical co-occurrence of TTP and SS is so rare that it might be easily neglected by physicians. As a life-threatening emergency, TTP requires prompt diagnosis and PET should be initiated as rapidly as possible. [12] We hope that this case can raise vigilance for TTP accompanied by autoimmune diseases in males.
Second, TTP preceded the diagnosis of SS in this case and no evidence of the other extra-glandular involvement was found after careful evaluation. In 5 out of 11 previously reported cases, SS was diagnosed later than the onset of TTP. A single-center study focused on autoimmune TTP also revealed that 17 out of 56 (30.4%) patients presented an autoimmune disorder after TTP [28]. In terms of SS, though ocular and oral dryness is the pivotal clinical pattern, patients may take little notice of it, just as what happened in this case. Actually, a great number of non-sicca symptoms might arise before dryness, such as fever of unknown origin, parotid gland swelling, and fatigue [8], especially in Asian population [30]. Incipient clinical presentations could also be extra-glandular involvement, including renal tubular acidosis, interstitial lung disease, cutaneous vasculitis, peripheral neuropathy, leukopenia, arthritis, primary biliary cirrhosis, Hashimoto’s thyroiditis, and TTP [4, 8, 34]. Therefore, it is challenging to recognize SS with subclinical sicca symptoms and autoantibody screening provides important clues. In as many as 66% of SS patients, autoantibodies (ANA, rheumatoid factor, anti-SSA, anti-SSB) appear years before symptom onset [16]. Additionally, it has been reported that the presence of positive anti-dsDNA or anti-SSA could be a risk factor for autoimmune disease development after TTP [28]. In this case, the positive autoantibody test motivated us to trace back the history and perform sicca evaluation, which finally led to the diagnosis of SS. In this way, our case highlights the necessity of autoantibody screening in defining underlying diseases of TTP.
Third, the patient had severe ADAMTS-13 activity deficiency (< 10%) [19] with detectable inhibitor in this case. Among the nine medical centers previously reporting SS-related TTP, only three of them measured ADAMTS-13 activity, reflecting the limited use of this test. Nonetheless, ADAMTS13 activity is highly valued in both diagnosis and prognosis prediction of TTP [40]; thus, its assessment should be strongly advocated. According to the recent epidemiological study, 75% of ADAMTS-13 activity deficiency is induced by anti-ADAMTS-13 IgG, which is potentially caused by underlying autoimmune diseases [21]. Since the patient in this case had no other concomitant autoimmune or hematological disease, the ADAMTS-13 inhibitor could be anti-ADAMTS-13 IgG associated with SS. It remains to be investigated why anti-ADAMTS-13 IgG is produced in SS. Inherent links lying in their pathophysiology might shed a light. Recent pre-clinical study revealed that the depletion of the precursor endothelial pool might occur in SS, hampering vascular endothelial restoration [5]. The resulted vascular endothelial injury participates in stimulating the release of vWF and aggravating the formation of microthrombi in TTP [10]. Based on these findings, we could speculate that TTP and SS are perhaps more closely related than currently believed.
Finally, treated by glucocorticoid and CTX, the patient experienced no relapse after PET discontinued. Among 11 previously reported cases, only four patients recovered without relapse. Undoubtedly, PET serves as the cornerstone of the treatment of TTP, despite the presence of SS [11]. However, as high as 40% of the autoimmune TTPs relapse after PET [33]. Furthermore, patients with ADAMTS-13 activity deficiency during remission are predicted to have even higher relapse risk [24]. For this reason, adjunctive immunosuppressive treatment is supposed to show great potential in preventing TTP relapse by eliminating anti-ADAMTS-13 antibodies [13], but large randomized controlled trails are still required. This case supported the efficacy of glucocorticoid and CTX. Besides, the monoclonal antibody against CD20 receptor, rituximab, has also demonstrated promise in reducing the frequency of TTP relapse [19, 25, 41].
Conclusion
In conclusion, we presented the first case of SS-associated TTP in male patients, which elucidated the importance of autoantibody screen in the workup of TTP and the benefits of adjunctive immunosuppressive therapy in relapse prevention.
References
Abe H, Tsuboi N, Yukawa S, Tsuji S, Hayashi H, Yukawa N, Takanashi H, Tahara K, Tonozuka N, Hayashi T (2004) Thrombotic thrombocytopenic purpura complicating Sjogren’s syndrome with crescentic glomerulonephritis and membranous nephritis. Mod Rheumatol 14(2):174–178. https://doi.org/10.1007/s10165-004-0287-4
Abu-Hishmeh M, Sattar A, Zarlasht F, Ramadan M, Abdel-Rahman A, Hinson S, Hwang C (2016) Systemic lupus erythematosus presenting as refractory thrombotic thrombocytopenic purpura: a diagnostic and management challenge. A case report and concise review of the literature. Am J Case Rep 17:782–787. 10.12659/AJCR.898955
Allan DS, Kovacs MJ, Clark WF (2001) Frequently relapsing thrombotic thrombocytopenic purpura treated with cytotoxic immunosuppressive therapy. Haematologica 86(8):844–850
Barone F, Colafrancesco S (2016) Sjogren’s syndrome: from pathogenesis to novel therapeutic targets. Clin Exp Rheumatol 34(4 Suppl 98):58–62
Bartoloni E, Alunno A, Bistoni O, Caterbi S, Luccioli F, Santoboni G, Mirabelli G, Cannarile F, Gerli R (2015) Characterization of circulating endothelial microparticles and endothelial progenitor cells in primary Sjogren’s syndrome: new markers of chronic endothelial damage? Rheumatology (Oxford) 54(3):536–544. https://doi.org/10.1093/rheumatology/keu320
Beloncle F, Buffet M, Coindre JP, Munoz-Bongrand N, Malot S, Pene F, Mira JP, Galicier L, Guidet B, Baudel JL, Subra JF, Tanguy-Schmidt A, Pourrat J, Azoulay E, Veyradier A, Coppo P (2012) Splenectomy and/or cyclophosphamide as salvage therapies in thrombotic thrombocytopenic purpura: the French TMA Reference Center experience. Transfusion 52(11):2436–2444. https://doi.org/10.1111/j.1537-2995.2012.03578.x
Brandt JE, Priori R, Valesini G, Fairweather D (2015) Sex differences in Sjogren’s syndrome: a comprehensive review of immune mechanisms. Biol Sex Differ 6(1):19. https://doi.org/10.1186/s13293-015-0037-7
Brito-Zeron P, Ramos-Casals M (2014) Advances in the understanding and treatment of systemic complications in Sjogren’s syndrome. Curr Opin Rheumatol 26(5):520–527. https://doi.org/10.1097/bor.0000000000000096
Campbell GN, Gallo JH (1998) Relapsing thrombotic thrombocytopenic purpura (TTP) in Sjogren’s syndrome. Aust NZ J Med 28(2):214. https://doi.org/10.1111/j.1445-5994.1998.tb02974.x
Chauhan AK, Goerge T, Schneider SW, Wagner DD (2007) Formation of platelet strings and microthrombi in the presence of ADAMTS-13 inhibitor does not require P-selectin or beta3 integrin. J Thromb Haemost 5(3):583–589. https://doi.org/10.1111/j.1538-7836.2006.02361.x
Coppo P (2017) Treatment of autoimmune thrombotic thrombocytopenic purpura in the more severe forms. Transfus Apher Sci 56(1):52–56. https://doi.org/10.1016/j.transci.2016.12.019
Coppo P, Veyradier A (2012) Current management and therapeutical perspectives in thrombotic thrombocytopenic purpura. Presse Med 41(3):e163–e176. https://doi.org/10.1016/j.lpm.2011.10.024
George JN (2012) Corticosteroids and rituximab as adjunctive treatments for thrombotic thrombocytopenic purpura. Am J Hematol 87(Suppl 1):S88–S91. https://doi.org/10.1002/ajh.23126
Guzicka-Kazimierczak R, Kazimierczak A, Clark J (2015) Promising results from cyclophosphamide based immunosuppression therapy of thrombotic thrombocytopenic purpura. Pomeranian J Life Sci 61(1):34–40
Hertzberg MS, Koutts J (1997) Oral cyclophosphamide for refractory or relapsing thrombotic thrombocytopenic purpura (TTP). Aust NZ J Med 27(4):439. https://doi.org/10.1111/j.1445-5994.1997.tb02204.x
Jonsson R, Theander E, Sjostrom B, Brokstad K, Henriksson G (2013) Autoantibodies present before symptom onset in primary Sjogren syndrome. JAMA 310(17):1854–1855. https://doi.org/10.1001/jama.2013.278448
Knobl P (2014) Inherited and acquired thrombotic thrombocytopenic purpura (TTP) in adults. Semin Thromb Hemost 40(04):493–502. https://doi.org/10.1055/s-0034-1376883
Koga T, Yamasaki S, Nakamura H, Kawakami A, Furusu A, Taguchi T, Eguchi K (2013) Renal thrombotic microangiopathies/thrombotic thrombocytopenic purpura in a patient with primary Sjogren’s syndrome complicated with IgM monoclonal gammopathy of undetermined significance. Rheumatol Int 33(1):227–230. https://doi.org/10.1007/s00296-010-1569-0
Lim W, Vesely SK, George JN (2015) The role of rituximab in the management of patients with acquired thrombotic thrombocytopenic purpura. Blood 125(10):1526–1531. https://doi.org/10.1182/blood-2014-10-559211
Lin TY, Chang CC, Chang CC, Yuan JY, Chen HH (2012) Cyclophosphamide-rescued plasmapheresis-unresponsive secondary thrombotic thrombocytopenic purpura caused by Sjogren’s syndrome. Arch Med Sci 8(5):934–938. https://doi.org/10.5114/aoms.2012.30788
Mariotte E, Azoulay E, Galicier L, Rondeau E, Zouiti F, Boisseau P, Poullin P, de Maistre E, Provot F, Delmas Y, Perez P, Benhamou Y, Stepanian A, Coppo P, Veyradier A (2016) Epidemiology and pathophysiology of adulthood-onset thrombotic microangiopathy with severe ADAMTS13 deficiency (thrombotic thrombocytopenic purpura): a cross-sectional analysis of the French national registry for thrombotic microangiopathy. Lancet Haematol 3(5):e237–e245. https://doi.org/10.1016/s2352-3026(16)30018-7
Murrin RJ, Murray JA (2006) Thrombotic thrombocytopenic purpura: aetiology, pathophysiology and treatment. Blood Rev 20(1):51–60. https://doi.org/10.1016/j.blre.2005.02.001
Noda M, Kitagawa M, Tomoda F, Iida H (1990) Thrombotic thrombocytopenic purpura as a complicating factor in a case of polymyositis and Sjogren’s syndrome. Am J Clin Pathol 94(2):217–221. https://doi.org/10.1093/ajcp/94.2.217
Page EE, Kremer Hovinga JA, Terrell DR, Vesely SK, George JN (2016) Clinical importance of ADAMTS13 activity during remission in patients with acquired thrombotic thrombocytopenic purpura. Blood 128(17):2175–2178. https://doi.org/10.1182/blood-2016-06-724161
Page EE, Kremer Hovinga JA, Terrell DR, Vesely SK, George JN (2016) Rituximab reduces risk for relapse in patients with thrombotic thrombocytopenic purpura. Blood 127(24):3092–3094. https://doi.org/10.1182/blood-2016-03-70382
Porola P, Laine M, Virkki L, Poduval P, Konttinen YT (2007) The influence of sex steroids on Sjogren’s syndrome. Ann N Y Acad Sci 1108(1):426–432. https://doi.org/10.1196/annals.1422.045
Porola P, Laine M, Virtanen I, Pollanen R, Przybyla BD, Konttinen YT (2010) Androgens and integrins in salivary glands in Sjogren’s syndrome. J Rheumatol 37(6):1181–1187. https://doi.org/10.3899/jrheum.091354
Roriz M, Landais M, Desprez J, Barbet C, Azoulay E, Galicier L, Wynckel A, Baudel JL, Provot F, Pene F, Mira JP, Presne C, Poullin P, Delmas Y, Kanouni T, Seguin A, Mousson C, Servais A, Bordessoule D, Perez P, Chauveau D, Veyradier A, Halimi JM, Hamidou M, Coppo P (2015) Risk factors for autoimmune diseases development after thrombotic thrombocytopenic purpura. Medicine (Baltimore) 94(42):e1598. https://doi.org/10.1097/md.0000000000001598
Sadler JE, Moake JL, Miyata T, George JN (2004) Recent advances in thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program 2004(1):407–423. https://doi.org/10.1182/asheducation-2004.1.407
Sandhya P, Jeyaseelan L, Scofield RH, Danda D (2015) Clinical characteristics and outcome of primary Sjogren’s syndrome: a large Asian Indian cohort. Open Rheumatol J 9(1):36–45. https://doi.org/10.2174/1874312901409010036
Sayani FA, Abrams CS (2015) How I treat refractory thrombotic thrombocytopenic purpura. Blood 125(25):3860–3867. https://doi.org/10.1182/blood-2014-11-551580
Schattner A, Friedman J, Klepfish A (2002) Thrombotic thrombocytopenic purpura as an initial presentation of primary Sjogren’s syndrome. Clin Rheumatol 21(1):57–59. https://doi.org/10.1007/s100670200013
Scully M, Cataland S, Coppo P, de la Rubia J, Friedman KD, Kremer Hovinga J, Lammle B, Matsumoto M, Pavenski K, Sadler E, Sarode R, Wu H (2017) Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost 15(2):312–322. https://doi.org/10.1111/jth.13571
Selva O'Callaghan A, Bosch Gil JA, Solans Laque R, Segura Garcia A, Armadans Gil L, Mijares Boeckh-Behrens T, Vilardell Tarres M (2001) Primary Sjogren’s syndrome: clinical and immunological characteristics of 114 patients. Med Clin (Barc) 116(19):721–725
Shenkman B, Einav Y (2014) Thrombotic thrombocytopenic purpura and other thrombotic microangiopathic hemolytic anemias: diagnosis and classification. Autoimmun Rev 13(4-5):584–586. https://doi.org/10.1016/j.autrev.2014.01.004
Shiboski CH, Shiboski SC, Seror R, Criswell LA, Labetoulle M, Lietman TM, Rasmussen A, Scofield H, Vitali C, Bowman SJ, Mariette X (2017) 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjogren’s syndrome: a consensus and data-driven methodology involving three international patient cohorts. Ann Rheum Dis 76(1):9–16. https://doi.org/10.1136/annrheumdis-2016-210571
Steinberg AD, Green WT Jr, Talal N (1971) Thrombotic thrombocytopenic purpura complicating Sjogren’s syndrome. JAMA 215(5):757–761. https://doi.org/10.1001/jama.1971.03180180033007
Toumeh A, Josh N, Narwal R, Assaly R (2014) Refractory thrombotic thrombocytopenic purpura associated with primary Sjogren syndrome treated with rituximab: a case report. Am J Ther 21(2):e56–e60. https://doi.org/10.1097/MJT.0b013e3182459aa0
Verbeke L, Delforge M, Dierickx D (2010) Current insight into thrombotic thrombocytopenic purpura. Blood Coagul Fibrinolysis 21(1):3–10. https://doi.org/10.1097/MBC.0b013e32833335eb
Wu N, Liu J, Yang S, Kellett ET, Cataland SR, Li H, HM W (2015) Diagnostic and prognostic values of ADAMTS13 activity measured during daily plasma exchange therapy in patients with acquired thrombotic thrombocytopenic purpura. Transfusion 55(1):18–24. https://doi.org/10.1111/trf.12762
Yamashita H, Takahashi Y, Kaneko H, Kano T, Mimori A (2013) Thrombotic thrombocytopenic purpura with an autoantibody to ADAMTS13 complicating Sjogren’s syndrome: two cases and a literature review. Mod Rheumatol 23(2):365–373. https://doi.org/10.1007/s10165-012-0644-7
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Consent
A written informed consent was signed by the patient concerning publication of this case report. A copy of this consent is available for editors of this journal.
Disclosures
None.
Rights and permissions
About this article

Cite this article
Xu, X., Zhu, T., Wu, D. et al. Sjögren’s syndrome initially presented as thrombotic thrombocytopenic purpura in a male patient: a case report and literature review. Clin Rheumatol 37, 1421–1426 (2018). https://doi.org/10.1007/s10067-017-3912-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-017-3912-2