
Abstract
Blau syndrome (BS) is a rare autosomal dominant autoinflammatory disease characterized by the clinical triad of dermatitis, arthritis, and uveitis. It is caused by mutations in nucleotide-binding oligomerization domain-containing protein-2 (NOD2) gene. BS has been widely reported in Caucasians but cases documented in China are scarce. We reported two Chinese families with BS, which were by far the two largest pedigrees in the Chinese population. We identified two unrelated families with BS. The phenotypes and genotypes of these patients were reviewed and compared with previous cohorts. The proband of the first family was a 32-year-old Chinese Han woman, who had dermatitis, polyarthritis, and intermittent fever since the age of 6, bilateral panuveitis since 12. During her disease course, she lost her vision and developed hand flexion contractures. The proband of the second family was a 36-year-old Chinese Han woman, who had dermatitis and bilateral panuveitis since the age of 7, persistent polyarthritis since 13. Additional 7 and 4 family members were affected in the first and second families, respectively, and pedigree analysis suggested autosomal dominant inheritance. Genetic testing in both families identified the heterozygous c.1000 C > T, R334W mutation in NOD2 gene. Only one patient had recurrent fever as an expanded manifestation beyond the classical triad. BS can occur in multiple ethnic groups including the Chinese Han population. Our 11 adult patients constituted the largest adult cohort of BS ever reported in China. Lack of recognition of BS led to a significant delay in diagnosis. A considerable percentage of patients did not demonstrate the full spectrum of the classical triad, further complicating the diagnosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Blau syndrome (BS) is a rare autosomal dominant, autoinflammatory granulomatous disorder [1], which is linked to certain variants of the nucleotide-binding oligomerization containing protein-2 (NOD2) gene [2]. The disease usually starts in early childhood with clinical features consisting of one or more of the following granulomatous inflammations: uveitis, arthritis (sometimes camptodactyly), and dermatitis.
The protein of NOD2 is a member of a family of intracellular proteins with N-terminal caspase recruitment domains (CARDs) [3, 4]. Since the discovery of NOD2 in 2001, NOD2 variants have been associated with Crohn’s disease, Blau syndrome (BS), and most recently, Yao syndrome (YAOS, formerly known as NOD2-associated autoinflammatory disease, NAID) [5, 6]. BS has been linked to specific NOD2 variants, R334W and R334Q being the most commonly observed [7].
BS has been widely reported in Caucasians but only sparsely documented in Asians [7, 8], especially in the Chinese population [9, 10]. In this study, we described two unrelated families of BS in China, including 13 affected members comprising 11 adults and 2 children, all harboring the R334W mutation in NOD2.
Patients and methods
Both probands were referred to our tertiary medical center, and complete medical records were documented, including the pedigree and the disease histories of kindreds. This research was approved by the Institutional Review Board of Peking Union Medical College Hospital and performed according to the Declaration of Helsinki. Informed consents were obtained from all participants. Whole-exome sequencing by next-generation sequencing was performed in the Center for Genetic Testing, Joy Orient Translational Medicine Research Centre Co., Ltd., Beijing, China.
Results
The first family
The proband of the first family (III:1, Fig. 1) was a 32-year-old Chinese Han woman. She had chronic polyarthritis involving bilateral metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints of the hands, wrists, elbows, knees, and ankles accompanied by papular rashes on four extremities since the age of 6. She also had intermittent fever since then. Disease-modifying anti-rheumatic drugs (DMARDs) such as methotrexate and leflunomide combined with low-dose prednisone had no effect. At 12 years of age, she developed persistent bilateral panuveitis, which was resistant to the combination of cyclosporine and corticosteroids. Over the years, her polyarthritis resulted in flexion contractures of her third, fourth, and fifth fingers of both hands (camptodactyly) (Fig. 2a) and persistent swelling of both knees, and her uveitis caused atrophy of both eyeballs and eventually complete loss of vision. The patient denied any symptoms suggestive of inflammatory bowel disease (IBD), such as prolonged diarrhea or abdominal pain, melena, hematochezia, or perianal lesions. She also denied manifestations more specific to YAOS, such as distal lower extremity swelling or sicca-like symptoms.

Pedigree of the first family. The arrow indicates the proband. The asterisks indicate the individuals who had undergone clinical and genetic analysis. Patients with Blau syndrome are marked in black and carriers of the NOD2 c.1000 C > T, R334W mutation are marked with a plus sign (+)

Phenotypes of the first family. Camptodactyly in the proband (a). X-ray of the proband showed symmetrical involvement of PIPs, joint space narrowing, subluxation, and contracture of multiple PIPs (b). Camptodactyly in other family members (II:1, II:3, IV:1) (c, d, e). Papules on the upper limb of the proband’s son (IV:1) (f)
Blood tests showed that the complete blood count (CBC), complete biochemistry panel, and serum angiotensin-converting enzyme were all within normal range. Urine analysis (sediment and dipstick) was normal. Fecal occult blood test was negative. Serological testing for autoimmune diseases, including anti-nuclear antibody, anti-extractable nuclear antibodies, anti-double-stranded DNA, anti-neutrophil cytoplasmic antibodies, rheumatoid factor, anti-cyclic citrullinated peptide antibodies were all negative. HLA-B27 was negative. The erythrocyte sedimentation rate (ESR) was 34 mm/h (normal range 0–20), high-sensitivity C-reactive protein (hsCRP) was 23.6 mg/L (normal range 0–3), and tumor necrosis factor (TNF) α was 114.0 pg/ml (normal range 0–8.1). Her chest radiograph showed no abnormality. X-ray of her hands revealed symmetrical involvement of PIPs with joint space narrowing, subluxation, and contracture (Fig. 2b). Biopsy of synovium or skin rash was not performed due to the patient’s preference. Genetic testing identified a heterozygous c.1000 C > T, R334W mutation in NOD2 gene.
Pedigree analysis (Fig. 1) showed that her maternal grandmother (I:1), who died at the age of 70, developed bilateral “eye inflammation” and deforming polyarthritis of the hands since early childhood and eventually became blind in her 40s. The proband’s mother (II:1), a 54-year-old woman, had polyarthritis at 3 years of age and flexion contractures of the third through fifth fingers (Fig. 2c) developed. One of the proband’s maternal uncles (II:3) who was 48 years old had the similar articular manifestations as the proband’s mother since the age of 4 (Fig. 2d). One of her maternal aunts (II:5), a 43-year-old woman, suffered from recurrent anterior uveitis, hand flexion contractures, and ankle swelling since the age of 5. The two brothers of the proband (III:2 and III:4), 27 and 25 years old, respectively, both developed intermittent polyarthritis with bilateral flexion contractures of the third through fifth fingers since the age of 6. The proband’s 8-year-old son suffered from recurrent polyarthritis and chronic papules on his four limbs at 6 months after birth, and gradually developed bilateral flexion of the fourth and fifth PIPs (Fig. 2e, f), without uveitis or fever. All her affected kindreds had no IBD symptoms. All living patients’ CBC and biochemistry panel were within normal range; urine analysis and fecal occult blood test were normal. Autoantibodies were all negative. All the patients had mild to moderate elevation in ESR (ranging from 32 to 66 mm/h) and hsCRP (ranging from 10 to 55 mg/L). Genetic analysis confirmed the presence of the NOD2 c.1000 C > T, R334W heterogeneous mutation in all the affected kindreds, while the mutation was not identified in other 7 healthy kindreds in her family.
The proband and her son were treated with infliximab plus methotrexate with satisfactory response for the polyarthritis. ESR and hsCRP rapidly decreased to normal levels after therapy. Due to personal financial constraints, other affected members only received DMARDs such as methotrexate or cyclosporine combined with low to moderate dose of prednisone for their arthritis or uveitis with very limited efficacy.
The second family
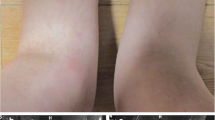
The proband of the second family (II:4, Fig. 3) was a 36-year-old Chinese Han woman. The patient had chronic bilateral panuveitis since the age of 7, which led to blindness 7 years later. She also suffered from widespread intermittent erythematous maculopapular rash without pruritus since the onset of the uveitis. She developed persistent polyarthritis since the age of 13, involving PIPs of the hands and bilateral wrists, elbows, knees (Fig. 4a), and ankles. The affected joints had prolonged morning stiffness, mild tenderness, but no obvious deformity. The patient only received irregular nonsteroidal anti-inflammatory drugs (NSAIDs) for her arthritis with partial relief. She had no IBD symptoms, distal lower extremity swelling, or sicca-like symptoms.

Pedigree of the second family. The arrow indicates the proband. The asterisks indicate the individuals who had undergone clinical and genetic analysis. Patients with Blau syndrome are marked in black and carriers of the NOD2 c.1000 C > T, R334W mutation are marked with a plus sign (+)

Periarticular boggy swelling of the second proband’s knees with mild inflammation (a). Ultrasonography of her left wrist showed effusion and marked synovial thickening (b). Ultrasonography of her right elbow showed a synovial cyst typical of Blau syndrome (c)
Blood tests for CBC, biochemistry panel, and autoantibodies were all normal. Urine analysis (sediment and dipstick) was normal, and the fecal occult blood test was negative. ESR was 48 mm/h, hsCRP was 33 mg/L, and TNFα was 10.1 pg/ml. Her chest radiograph was normal. X-ray of hands showed only soft tissue swelling and joint space narrowing of PIPs and wrists. Articular ultrasonography (Fig. 4b, c) revealed effusion, synovial cysts, and marked proliferation of the synovium in the afflicted joints. Synovium or skin biopsy was declined by the patient. Genetic testing identified a heterozygous c.1000 C > T, R334W mutation in NOD2 gene.
Pedigree analysis (Fig. 3) revealed that her 66-year-old mother (I:1) lost vision during childhood due to “chronic redness of eyes.” One of the proband’s sisters (II:2), 42 years old, had similar manifestations as the proband since the age of 5, including bilateral panuveitis leading to loss of vision, intermittent dermatitis, and chronic polyarthritis. Her 40-year-old brother (II:3) had intermittent anterior uveitis since the age of 4 and nondeforming polyarthritis since the age of 10, which was misdiagnosed as rheumatoid arthritis (RA). His 6-year-old daughter (III:2), the proband’s niece, developed widespread multiple papules, symmetric polyarthritis, and recurrent anterior uveitis since infancy. All her affected kindreds denied symptoms suggestive of IBD. Genetic analysis demonstrated NOD2 c.1000 C > T, R334W heterogeneous mutation in the 4 affected kindreds but not in other healthy members in this family.
The proband and her niece both received infliximab plus methotrexate with satisfactory responses for arthritis and dermatitis, and the uveitis of her niece also subsided. Other members only received traditional DMARDs combined with low to moderate dose of prednisone with marginal efficacy.
Discussion
BS is an autosomal dominant disease typically characterized by the clinical triad of granulomatous dermatitis, inflammatory arthritis, and uveitis. Although BS is reported predominantly among Caucasians, it has been reported in East Asia, including 34 cases in Japan [11], 19 cases in China [9, 10], and 4 cases in South Korea [12]. Since the majority of the aforementioned cases were sporadic, we herein described the two largest pedigrees of BS in East Asia as well as in China.
Both our probands presented with typical “boggy” arthritis, panuveitis, and papular rashes. Their diagnoses as BS were confirmed by the presence of a common NOD2 mutation in BS and a family history of autosomal dominant inheritance. The median age of our patients at diagnosis was 40 (ranging from 6 to 70), which was much older than the previous cohorts [7, 10, 13, 14] mainly due to the decades of delay in diagnosis. Our 11 adult patients constituted the largest adult cohort of BS in China. The first symptom(s) of all patients except two probands began during early childhood, which was consistent with previous reports.
The phenotypic and genotypic characteristics of our cohort were summarized in Table 1, along with previously reported Chinese cohorts [9, 10] and recently published large cohorts [7, 13, 14]. As in other cohorts, arthritis was the most common component among the classical clinical triad, with a prevalence of 92%. Despite sharing the same NOD2 mutation, all the 8 patients from the first family developed camptodactyly and hand flexion contractures, whereas none from the second family had joint deformity. This marked discrepancy suggested other genetic or nongenetic factors might be involved in phenotypic variation of BS. In our cohort, 8 patients suffered from uveitis, resulting in complete visual loss in 5 (38%). The severity of ocular outcome highlights the importance of early recognition and proper management of BS. Only 38% of our patients suffered from dermatitis, which was significantly less frequent than the Japanese cohort and the Caucasian cohorts [7, 13, 14]. Since skin involvement in BS could be transient and asymptomatic, the retrospective nature of our study and long disease courses of our patients might contribute to this difference. Nevertheless, skin rashes of our patients manifested mostly as multiple papules just as reported in the previous cohorts. In general, the severity of each individual component of the classical triad varied considerably from case to case even in the same pedigree, suggesting heterogeneity of phenotype of BS. Hence, our cohort and other studies show BS as an autosomal dominant trait with varying expressivity [8, 11, 13, 14].
During the disease course, fewer patients (31%) in our cohort developed the complete clinical triad of dermatitis, arthritis, and uveitis, compared with the previous large cohorts (71–85%) [13, 14]. This difference might be partly explained by the fact that our cohort consisted entirely of familial cases whose diagnosis depended more on intentional gene screening rather than on typical clinical features. Whereas, the previous cohorts included 51–70% sporadic cases, and the diagnosis of sporadic cases relies heavily on the presence of classical triad to prompt gene testing. Surprisingly, as many as 39% patients in our cohort developed only one component of the classical triad, emphasizing the importance of family history in the differential diagnosis of fairly common symptoms. Therefore, BS should be considered in children presenting with even one component of the classical triad of arthritis, dermatitis, and uveitis.
Expanded manifestations beyond the classical triad have been reported [7, 13, 14], such as recurrent or persistent fever, cutaneous vasculitis, panniculitis, granulomatous lymphadenopathy, pneumonitis, hepatosplenomegaly, granulomatous glomerulonephritis and interstitial nephritis, hypertension, cerebral infarction, cranial neuropathies, and large-vessel vasculitis. Among our patients, no history of the above-mentioned manifestations was documented except one patient (the first proband) with recurrent fever. Physical examinations revealed no hypertension, lymphadenopathy, or hepatosplenomegaly. Urine analysis and biochemistry panel all showed normal results. Hence, the frequency of expanded features among our patients was only 8%, compared with the frequencies of 25–75% in other cohorts [7, 13, 14]. The reason for this discrepancy was probably twofold. First, due to lack of thoracic CT scan, cranial MRI and large-artery ultrasonography or CT angiography, the frequency of asymptomatic involvement of lung, central nervous system, or large-vessel vasculitis might be underestimated; second, it had been suggested that patients with R334W mutation might have less expanded symptoms than those with other mutations [13]. However, the literature [7, 10, 13, 14] demonstrated that a significant percentage (13–55%) of BS patients had fever (recurrent or persistent) as an expanded feature, indicating that BS should be included in the differential diagnosis of fever of unknown origin in children.
BS has been linked to specific NOD2 sequence variants, including R334W, R334Q, E383K, E383G, G464W, L469F, W490L, M513R, R587C, T605N, H496L, M513T, C495Y, and N670K. The two most frequent mutations are R334W and R334Q [2], found in 60–80% patients when taken together [7, 13, 14]. The NOD2 sequence variants in BS are primarily located in the nucleotide-binding domain (NBD) region, and some of these mutants have “gain of function” in vitro studies [2]. Each patient is heterozygous for a specific mutation, and rare asymptomatic carriers had been described [13]. Our patients, along with the other Chinese BS patients [9, 10], demonstrated R334W variant as the most common mutation (56%) in the Chinese population, similar to the Japanese cohort (45%) [14]. More studies are needed to further clarify the genotype of BS in China. Crohn’s disease and YAOS have also been associated with NOD2 gene, but with different mutations (1007fs, G908R, R702W for Crohn’s disease, and IVS8+158, R702W for YAOS) [5, 6]. Since our patients had a mutation very specific to BS without any symptoms more specific to IBD or YAOS, the latter two diagnoses could be ruled out with confidence.
In terms of treatment, all of our patients received DMARDs and/or low to moderate dose of prednisone before the diagnosis of BS. Among patients with predominant arthritis, methotrexate was the most commonly used DMARD, whereas cyclosporine was often given to patients with chronic uveitis. Despite their long-term treatment, most patients remained symptomatic with progressive worsening of their hand flexion contractures and/or vision loss. All patients had mild to moderate elevation in ESR and hsCRP, which were also indications for their persistent disease activity. After the diagnosis of BS, 4 patients received infliximab plus methotrexate with satisfactory clinical response and normalization of ESR and hsCRP. TNFα inhibitors such as infliximab are the most commonly used biological therapy in BS [1, 7], and the superiority over traditional DMARDs has also been documented [7, 11]. But the long-term efficacy of anti-TNFα agents, particularly with regard to ocular morbidity, remains unknown.
In conclusion, we reported the two largest pedigrees of BS in China, including the largest Chinese adult cohort. The older age of our patients at diagnosis illustrated unrecognition of BS in China. The large percentage of our patients who had only one component of the classical triad underlined the difficulty in entertaining the diagnosis of BS in clinical practice.
References
Wouters CH, Maes A, Foley KP, Bertin J, Rosé CD (2014) Blau syndrome, the prototypic auto-inflammatory granulomatous disease. Pediatr Rheumatol Online J 12:33
Sfriso P, Caso F, Tognon S, Galozzi P, Gava A, Punzi L (2012) Blau syndrome, clinical and genetic aspects. Autoimmun Rev 12(1):44–51
Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G (2001) Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem 276(7):4812–4818
McGovern DP, van Heel DA, Ahmad T, Jewell DP (2001) NOD2 (CARD15), the first susceptibility gene for Crohn’s disease. Gut 49(6):752–754
Yao Q (2013) Nucleotide-binding oligomerization domain containing 2: structure, function, and diseases. Semin Arthritis Rheum 43(1):125–130
Yao Q, Shen M, McDonald C, Lacbawan F, Moran R, Shen B (2015) NOD2-associated autoinflammatory disease: a large cohort study. Rheumatology (Oxford) 54(10):1904–1912
Rosé CD, Pans S, Casteels I, Anton J, Bader-Meunier B, Brissaud P et al (2015) Blau syndrome: cross-sectional data from a multicentre study of clinical, radiological and functional outcomes. Rheumatology (Oxford) 54(6):1008–1016
Wang X, Kuivaniemi H, Bonavita G, Mutkus L, Mau U, Blau E et al (2002) CARD15 mutations in familial granulomatosis syndromes: a study of the original Blau syndrome kindred and other families with large-vessel arteritis and cranial neuropathy. Arthritis Rheum 46(11):3041–3045
Xiang H, Zhang T, Chen M, Zhou X, Li Z, Yan N et al (2012) NOD2/CARD15 gene mutation identified in a Chinese family with Blau syndrome. Mol Vis 18:617–623
Wang W, Wei M, Song H, Qiu Z (2014) Mutations of NOD2 gene and clinical features in Chinese Blau syndrome patients. Zhonghua Er Ke Za Zhi 52(12):896–901 Chinese
Otsubo Y, Okafuji I, Shimizu T, Nonaka F, Ikeda K, Eguchi K (2017) A long-term follow-up of Japanese mother and her daughter with Blau syndrome: effective treatment of anti-TNF inhibitors and useful diagnostic tool of joint ultrasound examination. Mod Rheumatol 27(1):169–173
Kim W, Park E, Ahn YH, Lee JM, Kang HG, Kim BJ, Ha IS et al (2016) A familial case of Blau syndrome caused by a novel NOD2 genetic mutation. Korean J Pediatr 59(Suppl 1):S5–S9
Rosé CD, Aróstegui JI, Martin TM, Espada G, Scalzi L, Yagüe J et al (2009) NOD2-associated pediatric granulomatous arthritis, an expanding phenotype: study of an international registry and a national cohort in Spain. Arthritis Rheum 60(6):1797–1803
Okafuji I, Nishikomori R, Kanazawa N, Kambe N, Fujisawa A, Yamazaki S et al (2009) Role of the NOD2 genotype in the clinical phenotype of Blau syndrome and early-onset sarcoidosis. Arthritis Rheum 60(1):242–250
Acknowledgements
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosures
None.
Funding
This work was supported by the National Natural Science Foundation of China (Grant no. 81501405) and the Youth Research Funds of Peking Union Medical College (Grant no. 3332015092).
Rights and permissions
About this article

Cite this article
Wu, D., Shen, M. Two Chinese pedigrees of Blau syndrome with thirteen affected members. Clin Rheumatol 37, 265–270 (2018). https://doi.org/10.1007/s10067-017-3758-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-017-3758-7