
Abstract
Background
The ethnic diversity of India provides a unique opportunity to study the history of the origin of mutations of genetic disorders. Spinocerebellar ataxia type 27B (SCA27B), a recently identified dominantly inherited cerebellar disorder is caused by GAA-repeat expansions in intron 1 of Fibroblast Growth Factor 14 (FGF14). Predominantly reported in the European population, we aimed to screen this mutation and study the founder haplotype of SCA27B in Indian ataxia patients.
Methods
We have undertaken screening of GAA repeats in a large Indian cohort of ~ 1400 uncharacterised ataxia patients and kindreds and long-read sequencing-based GAA repeat length assessment. High throughput genotyping-based haplotype analysis was also performed. We utilized ~ 1000 Indian genomes to study the GAA at-risk expansion alleles.
Findings
We report a high frequency of 1.83% (n = 23) of SCA27B in the uncharacterized Indian ataxia cohort. We observed several biallelic GAA expansion mutations (n = 5) with younger disease onset. We observed a risk haplotype (AATCCGTGG) flanking the FGF14-GAA locus over a 74 kb region in linkage disequilibrium. We further studied the frequency of this risk haplotype across diverse geographical population groups. The highest prevalence of the risk haplotype was observed in the European population (29.9%) followed by Indians (21.5%). The observed risk haplotype has existed through ~ 1100 generations (~ 22,000 years), assuming a correlated genealogy.
Interpretation
This study provides valuable insights into SCA27B and its Upper Paleolithic origin in the Indian subcontinent. The high occurrence of biallelic expansion is probably relevant to the endogamous nature of the Indian population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Spinocerebellar ataxia (SCA) is a class of genetically heterogeneous, progressive, neurodegenerative disorders inherited in an autosomal dominant fashion with clinical features of poor coordination and balance, cognitive impairment, nystagmus, and slurred speech. Thus far, many genotypically distinct SCAs have been recognized with overlapping yet distinct phenotypes with varied age-at-onset. Most of these SCAs are brought about by tandem repeat expansion in explicit loci (e.g., SCA1, SCA2, SCA3, and SCA6) while others occur due to deleterious mutations. Yet in this SCA heterogeneity with a prevalence of ~ 2.7 in 100,000 individuals, an ample number of cases with ataxia-like phenotype remain genetically undiagnosed. Only ~ 10–30% of cases clinically diagnosed with ataxia are identified with definitive genetic etiology while the larger proportion remains unresolved [1]. Recently, to overcome this challenge, a novel attempt to investigate further yielded a unique pathogenic repeat expansion identified to be associated with another progressive ataxia, SCA27B [2].
SCA27B is a late-onset, autosomal dominant repeat expansion disorder with tandem triplet (GAA) repeat expansion in intron 1 of FGF14 gene on chromosome 13. FGF14 belongs to the fibroblast growth factor (FGF) family that includes 22 proteins involved in regulating several physiological processes in both developing and adult individuals. During embryogenesis, FGFs play a crucial role in mitogenesis, cell migration, differentiation, and morphogenesis while in adults, they are involved in angiogenesis and tissue repair. Several members of the FGF family have been previously established as the underlying cause of different human disorders, namely, idiopathic hypogonadotropic hypogonadism (FGF8), colorectal, endometrial and ovarian carcinoma (FGF9), SCA27 (FGF14), and Parkinson disease (FGF20) [3]. The FGF14 gene is highly expressed in the brain, especially in Purkinje cells where it regulates neuronal excitability by interacting with voltage-gated channels. Additionally, FGF14 contributes to synaptic plasticity and neurogenesis in the hippocampus. Mutations or dysregulation of the FGF14 gene are associated with conditions such as epilepsy, ataxia, and cognitive impairment. Various missense [4], non-sense [5], deletion [6], and translocation [7] mutations in FGF14 gene have been well-established as the root cause of SCA27. The FGF14 gene demonstrates evident allelic heterogeneity, as different mutations within the same gene give rise to distinct disorders, namely, SCA27 and SCA27B. SCA27 is characterized by early-onset symptoms resulting from a loss-of-function point mutation in the FGF14 gene whereas SCA27B is a late-onset disorder caused by repeat expansion, leading to haploinsufficiency. In addition to the typical ataxia symptoms, SCA27 harbors other noticeable phenotypes including widespread tremor, orofacial dyskinesias, and psychiatric symptoms with a high degree of severity [8, 9]. However, there have been no documented cases of SCA27B with such severe phenotypes, and individuals with SCA27B commonly exhibit the typical ataxia symptoms without this additional severe manifestations. The identification of this pathogenic GAA repeat expansion (GAA > 250) in FGF14 causing SCA27B has paved new opportunities to analyze the unsolved ataxia cases altogether retrospectively.
In the last two decades in India, with persistent effort and commitment, multiple scholars and research groups have deciphered the prevalence of SCA in our population as well as different ethnicities. Among the subtypes of SCA, the most prevalent is SCA12 which is otherwise rare worldwide. Other than that, SCA2 and SCA1 are the next most common forms of SCA diagnosed in the Indian population [10]. Previously, a study on 31 Indian index patients highlighted a notable prevalence (10%) of SCA27B in the Indian cohort [11]. In this paper, we have studied the prevalence of SCA27B over a large Indian cohort (n = 1402) to understand the frequency of this newly identified disorder in the Indian subcontinent.
Methodology
Cohort enrollment and genomic DNA extraction
To assess the prevalence of SCA27B in the Indian population, we enrolled a total of 1402 participants ranging in age from 4 months to 83 years. Of these 1402 participants, 1256 participants were index patients manifesting consecutive degenerative ataxia symptoms while 146 were kindreds. These participants were selected primarily from the All India Institute of Medical Science (AIIMS) in New Delhi, along with other reputable tertiary referral centres across the country, as part of the GOMED (Genomics and Other Omics Tools for Enabling Medical Decision) program. Skilled neurologists previously assessed the participants and they underwent screening for the traditional ataxias prevalent in the Indian population, specifically SCA1, SCA2, SCA3, SCA6, SCA7, SCA12, and SCA17 and Friedreich ataxia (FRDA) [10]. Notably, the participants recruited for this particular study on SCA27B had remained without a definitive etiological diagnosis following the initial SCA screening. A control cohort of 86 neurologically healthy individuals was also recruited to understand the polymorphism in FGF14 GAA repeat number in the Indian population.
All individuals involved in this research study gave their informed consent to participate, and ethical approval was obtained from the Institutional Human Ethics Committee of CSIR-IGIB. Genomic DNA was extracted from peripheral venous blood samples using the salting-out technique [12].
Genomic Screening of the SCA27B repeat
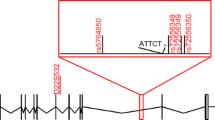
To screen the enlisted cohort for FGF14-GAA repeat expansion, the intronic repeat locus of FGF14 was amplified using primers flanking the GAA repeat region of FGF14 gene. The fluorescently labelled polymerase chain reaction (PCR) amplified product was evaluated for the number of repeat units by capillary electrophoresis on Genetic Analyzer 3500 Dx (Applied Biosystems, Thermo Fishers Scientific), and size estimation was done using GeneMapper V.6.0. PCR amplified products demonstrating only one visible allele peak or allelic peaks ≥ 550 bp in the capillary electrophoresis-derived electropherogram were further screened to ascertain repeat expansion through repeat-primed PCR. The samples demonstrating a repeat expansion in repeat-primed PCR were further evaluated for the number of repeating units through long-range PCR and estimated manually through 1% agarose gel electrophoresis. GAA repeat expansion ≥ 250 repeat units was considered as the pathogenic threshold [11]. The repeat motif at the short tandem repeat (STR) locus of patient samples presenting large amplification products beyond the pathogenic threshold was investigated through targeted long-read nanopore sequencing.
Amplicon-based long-read nanopore sequencing of GAA-FGF14 locus
We processed 24 samples, comprising 21 SCA27B positive samples and 3 exhibiting an interrupted RP-PCR sawtooth profile. The Oxford Nanopore Technology (ONT) library preparation followed an in-house protocol consistent with prior experiments [13]. Around 500 ng of the purified final libraries were loaded onto an ONT-MinION flow cell (R9.4.1) and sequenced on an ONT-MinION Mk1C device for a duration of around 20 h.
Following the conventional alignment of nanopore reads using Minimap2 [14] to reference region (GRCh37), bam files and raw intensity data (Fast5 files) were processed through STRique [15], a tool designed to quantify Short Tandem Repeats by localisation of repeat boundaries and hidden-Markov-based repeat counting. STRique output was filtered with custom parameters (suffix and prefix > = 4) to retain reads upstream and downstream primer sequences. Further, we applied a Gaussian Mixture Model (GMM) from the R Bioconductor package, Mclust [16], to separate reads based on the length of the repeat. This approach facilitates the identification of repeat lengths within each cluster, offering a more refined understanding of the STR landscape in each allele. To obtain the number of repeats in each allele for every sample, the following parameters were taken into consideration: (i) the total number of reads clustering together with preference to clusters with the highest number of reads; (ii) the frequency of the mode repeat in the cluster; (iii) the mode repeat of each cluster with relevance to LR-PCR data.
The consensus sequences were generated to study and validate: (i) all the SCA27B-positive samples carrying one or more expanded alleles are comprised of pure GAA-repeats; (ii) scan for possible interrupting motifs in samples with an interrupted profile in RP-PCR. The reads were then extracted from the selected clusters based on ± 20 repeats flanking the mode repeat sequence in fasta format to create a consensus fasta for each allele using Alfred [17] and checked for interruptions in repeats using Tandem Repeat Finder [18].
Repeat genotyping from an Indian genome dataset
To profile repeats in the Indian control population, we used ExpansionHunter [19], a sequence-graph-based tool for analyzing genome-wide STR profiling. We extracted repeat numbers from the IndiGen [20] dataset (n = 1014 samples), a consortium of 1000 Indian genomes, targeting the FGF14 repeat loci (hg38:chr13:102161575–102161726) which were further used in the haplotype analysis.
Haplotyping and Linkage Disequilibrium (LD) analysis
All the SCA27B positive samples (n = 23) were further genotyped using the Infinium Global Screening BeadChip Array-24 v3.0 (catalogue: 20031595) as per the manufacturer’s protocol. We analysed the data using GenomeStudio v2.1 to process the idat files.
We applied a Gentrain Score > = 0.7 and a cluster separation score > = 0.3 to filter out the bad-quality SNPs. All the samples passed the threshold of call rate > = 0.95. For downstream PLINK [21] processing, ~ 61,000 SNPs were taken from 23 samples. We filtered 41 SNPs with a Minor allele frequency (MAF) > = 0.05 from 200 kb upstream and downstream spanning the repeat region from the cases. We also extracted the same 41 SNPs from IndiGen using PLINK v1.9.With PHASE v2.1.1 [22], we used 100 iterations, 10 thinning intervals, and 100 burn-in iterations to calculate the Haplotype. We used the same cut-off values on PHASE to calculate from different populations available in the 1000 genome data for the same number of SNPs to check the frequency of the haplotypes in different world populations. We further used Pop-ART v1.7.2 [23] to visualise the distribution and differences in the various haplotypes across diverse population groups like Indian and 1000 genome populations i.e., African, American, South-Asian, East-Asian, and European.
To calculate the LD in IndiGen and 1000 genome population data, we used Haploview 4.1 [24], a software designed to visualize and analyse patterns of LD in genomic data.
Haplotype dating
The age of the mutation was determined using a published Haplotype dating method [25] and a website developed in the Bahlo Lab [https://shiny.wehi.edu.au/rafehi.h/mutation-dating/]. This method estimates the age of a genetic mutation based on the genetic length of ancestral haplotypes shared between individuals carrying the mutation. This approach provides the age of mutation and confidence intervals independent of the asymptotic theory and can be applied to genealogies with independent or correlated data.
Statistical analysis
In the genetically uncharacterized cases, the pre-mutable (large) normal allele cut-off was designated by taking observations under the two degrees of standard deviation (5%) from the mean of repeat lengths.
Results
Genotyping FGF14-GAA locus
We have studied the occurrence of SCA27B in a large Indian cohort of 1402 (1256 genetically unsolved ataxia patients; 146 kindreds) individuals with a mean age of 41 ± 18.4 years. Among the 1402 participants, 67% (n = 939) were male while the remaining 33% (n = 463) were female.
Initial screening of all 1402 samples by amplifying the GAA-repeat locus revealed the heterogeneity in the repeat lengths. 323 samples showing no or one allelic peak in the electropherogram underwent repeat-primed (RP) PCR. 75 samples demonstrating a saw-tooth electropherogram profile in RP-PCR underwent long-range (LR) PCR to determine the approximate length of the allele carrying the expanded repeating unit. The samples with repeats > 250 repeating units were processed for long-read nanopore sequencing. The data obtained from STRique followed a bimodal distribution pattern for each sample with two distinct clusters of reads. Hence, we adopted GMM for a better understanding of the STR landscape for each allele across every sample. We obtained multiple clusters for each sample following an unsupervised approach using MClust, R Bioconductor package. With this approach, we were able to obtain the allelic repeat data for 21 out of 24 samples processed in Nanopore (Fig. 1). The remaining 3 samples had low read count and read quality post-sequencing and STRique analysis. In comparison to long-read sequencing analysis, the repeat numbers calculated manually from LR-PCR had a standard deviation of ± 10 repeats for each allele across samples.

1 FGF14-GAA repeat distribution across cohort categories. The jitter-box plot shows the repeat distribution in each allele of every sample in the cohort. The three categories along the X-axis, namely, Control, Uncharacterised patients (unsolved SCA cases), and SCA27B (SCA27B positive cases) have been further subdivided based on alleles. Allele 1 (A1) and Allele (A2) have been given separate colours as mentioned in the figure legend. The Y-axis denotes the number of GAA repeats
In this paper, we report a notable frequency of 1.83% (n = 23/1256) for GAA repeat expansion in intron 1 of FGF14 gene corresponding to SCA27B. These individuals had repeat expansion in at least one allele beyond the pathogenic range of 250 GAA repeats extending from 262 to 645 repeats (mean GAA repeats: 338 ± 81). 21.7% (n = 5) of the SCA27B patients had a biallelic expansion while 8.6% (n = 2) of the patients had a homozygous repeat expansion. The heterogeneity at this locus was marked by successive repeating units from 6 to 645 with mode repeats as 10 repeating units. In the remaining genetically uncharacterized cases, the repeats varied between 6 and 245 repeats (mean GAA repeats: 24 ± 28). The pre-mutable (large) normal allele ranges between 80 and 250 as per the two degrees of standard deviations (5%) method. The estimated FGF14-GAA repeating units from our entire cohort with 2804 alleles reveal that 94.4% (n = 2648) were normal alleles (< 80 repeating units), 4.6% (n = 128) were large normal/intermediate alleles (80–250 repeating units), and 1% (n = 28) were expanded alleles (> 250 repeating units). The premutable normal allele range among various SCAs obtained from a previous study from our lab [10] was compared to that of SCA27B and has been provided in Supplementary Table 1. Among the samples with an interrupted RP-PCR profile, one demonstrated a prominent (GAAGCA)60 hexanucleotide repeat expansion associated with the interruptive profile. The remaining two samples had low read counts, hence, the interruptive motif could not be determined.
We conducted a study on the variation in the intronic GAA repeat locus of the FGF14 gene in a group of 86 neurologically healthy individuals serving as controls. These individuals varied in age between 1 year to 66 years (mean: 22 ± 19 years) and 59.3% (n = 51) were male while the remaining 40.7% (n = 35) were female. We further estimated the FGF14-GAA repeating unit from the Indigen dataset of 1014 samples. The estimated FGF14-GAA repeating units varied between 6 and 223 repeating units (mean GAA repeats: 26 ± 29) with a mode repeat of 10 repeating units. The entire control cohort with 2200 alleles reveals that 92% (n = 2024) were normal alleles (< 80 repeating units) while 8% (n = 176) were large normal/intermediate alleles (80–250 repeating units). The entire allelic distribution across each cohort is depicted in Fig. 2.

Distribution of repeats in all the reads for each sample obtained from STRique. The heat map demonstrates the distribution of reads for each sample across the entire repeat lengths. This helps us understand the distribution of alleles in each sample. The reads clustering together with a z-score above 0 indicate the allelic landscape corresponding to each sample. The X-axis denotes the GAA-repeat number while the Y-axis indicates the SCA27B positive sample IDs.
Clinical hallmarks of SCA27B patients
The clinical data revealed that the patients identified positive for SCA27B GAA repeat expansion varied in age from 17 to 79 years (mean = 51.2 ± 16.6 years). The frequency of occurrence of the disease in males and females is 82.6% (n = 19) and 17.3% (n = 4), respectively.
We obtained deeper insights into the clinical data of 15 out of 23 positive patient samples with varying age-at-onset between 14 and 71 years (mean = 50.2 ± 17.22 years). Among these, 80% (n = 12) individuals reported a negative family history indicating a de novo expansion while 20% (n = 3) individuals reported a relevant family history in one of the parents indicating the autosomal dominant nature of the disorder. The brain MRI of these individuals revealed diffused cerebral and cerebellar atrophy. A steady disease progression was observed in these individuals with a mean disease duration of 3.5 ± 2.4 years. The most common clinical features observed in these individuals included abnormal gait, cerebellar dysarthria, nystagmus, oculomotor apraxia i.e., broken ocular pursuits, and slow saccades. Few individuals reported pyramidal and extrapyramidal features. Two of the individuals reported autonomic dysfunctions such as urine incontinence and dysphagia.
Linkage Disequilibrium at the SCA27B locus
We studied LD localizing the GAA repeat motif of FGF14 gene across diverse geographical populations utilizing data from the 1000 Genomes Consortium and the IndiGen project. A total of 41 single nucleotide polymorphisms (SNPs) flanking 200 kb upstream and downstream of the repeat region were considered during haplotyping and LD analysis. For each population, the mean D’ (D’mean), mean LOD (LODmean), mean r2 value (r2mean), and confidence interval (CI) were calculated.
This analysis identified a prominent LD block encompassing the region of interest with 9 SNPs spanning over 74 kb. This LD block remains stable in South-Asian (D’mean=0.92; LODmean=45.5; r2mean = 0.31; CI = 0.83–0.95) and Indian (D’mean=0.86; LODmean=80.4; r2mean = 0.28; CI = 0.79–0.91) populations, while experiencing partial decay in other populations. The length of the LD block varies across groups, such as 67 kb in East-Asian (D’mean=0.85; LODmean=42.7; r2mean = 0.28; CI = 0.73–0.92), 60 kb in American (D’mean=0.84; LODmean=19.5; r2mean = 0.21; CI = 0.63–0.92), and 44 kb in African (D’mean=0.83; LODmean=17.7; r2mean = 0.1; CI = 0.55–0.9) populations. Notably, in the European (D’mean=0.73; LODmean=23.7; r2mean = 0.25; CI = 0.56–0.83) population, the region of interest is almost in linkage equilibrium, lacking a prominent LD block.
Insights on the haplotype landscape of SCA27B
Out of 2060 alleles (IndiGen = 2014, Patient = 46), 28 alleles had expansion over 250 repeats, 170 alleles lay in the intermediate range of 80–250 while the remaining 1862 were normal repeat alleles. Following PHASE v2.1.1, 42 unique haplotypes were perceived for this 74 kb stretch among these 2060 alleles. In these 28 expanded alleles, strikingly, 75% (n = 21) of the expanded allele shared a common haplotype i.e., AATCCGTGG (Haplo-1), 17.9% (n = 5) shared another haplotype i.e., AGCCCGTGG (Haplo-2) while the remaining 2 alleles had haplotypes AGTCCGTGG and AGCCTGTGG respectively. Similarly, in the 170 alleles with intermediate repeats, the most common haplotypes were the same as that of expanded alleles i.e., Haplo-1 and Haplo-2 accounting for 77.1% (n = 131) and 7.1% (n = 12) of the alleles respectively. In contrast, the most frequent haplotype in normal alleles was AGCCTGTGA (Haplo-7) acquitting 24.1% (n = 449) of the alleles which are otherwise absent in expanded alleles or infrequent in intermediate alleles (n = 1). The most common haplotypes of expanded and intermediate alleles i.e., Haplo-1 and Haplo-2 combinatorically accounted for only 16.2% (n = 302) of the normal alleles. In conclusion, Haplo-1 and Haplo-2 conjointly justify 92.9%, 84.1%, and 16.2% of the expanded, intermediate, and normal alleles respectively. This analysis is indicative that Haplo-1 may be a prominent risk haplotype with its major prevalence in the intermediate and expanded alleles. The association of the distinct haplotypes to various allele groups was assessed using a chi-square statistic test, yielding a value of 12.2682. The corresponding p-value is 0.002168, indicating statistical significance at the threshold of p < 0.05.
In the diverse population groups, the European population exhibits the highest prevalence of the risk haplotype at 29.9%, followed by 21.1% in Indian populations, 19.5% in South-Asian populations, 14.5% in African populations, and 7.6% in American populations as shown in Fig. 3.

A LD distribution spanning the repeat locus in diverse geographical populations. This heatmap for individual population groups (African, American, South-Asian, East-Asian, Indian, and European) was obtained from Haploview 4.1 pointing out the span of the LD block in each population. The red arrow marks the region of the FGF14 repeat locus. The rsIDs of each SNP are marked along the X-axis and the intensity of the red colour denotes the D’ value with a darker shade denoting D’ value closer to 1. B Haplotype analysis of the 74.5kb LD region among diverse geographical population. This analysis shows the distribution of 20 unique haplotypes among different population groups (African, American, South-Asian, East-Asian, Indian, and European). The figure legend represents the distinct colour assigned to each population group.
Origin of SCA27B repeat expansion mutation
We used the available SNP data from the patients to investigate the possibility of a ubiquitous haplotype underlying the expansion through generations. The core haplotype among the expanded and intermediate alleles is located at hg38:chr13:102096576–102171079 encompassing the FGF14-repeat motif and is 74 kb in size. Assuming a correlated genealogy, the mutation arose 1104.5 generations (CI-0.95: 680.2-1803.1) ago. Considering a 20-year generation span, the most recent common ancestor with this haplotype would have lived 22,100 years (CI-0.95: 13600-36060 13,600–36,060) ago. Considering a 25-year generation span, the most recent common ancestor with this haplotype would have lived 27,625 years (CI-0.95: 17,000–45,075) ago.
Discussion
Spinocerebellar ataxia is an autosomal dominant, progressive, genotypically heterogeneous class of neurodegenerative disorders. It presents abnormal gait and impairment of other cerebellar functions corresponding to debilitating effects on the individual. SCA27B is a late-onset, autosomal dominant disorder with tandem triplet (GAA) repeat expansion in intron 1 of FGF14 gene on chromosome 13 leading to haploinsufficiency [2].
This study provides valuable insights into the precise occurrence rate of SCA27B in a substantial cohort of ~ 1250 genetically uncharacterized ataxia patients relevant to the Indian context. In a previously reported study involving Indian index patients (n = 31), the disorder was indicated with a frequency of 10% [11]. However, this might be misleading when considering the overall uncharacterized ataxia cohort in which 1.83% accounts for this disorder. We identified 23 (82.6% male and 17.3% female) individuals with a mean age-at-onset of 50.2 ± 17.2 years with an expansion in at least one allele beyond the pathogenic range of 250 GAA repeat units. In the genetic spectrum of SCAs in the Indian population, SCA27B emerges as a strong candidate locus as its frequency is 1.83% which is higher than the previously determined frequencies of SCA6 (0.1%), SCA 7 (0.5%), and SCA 17 (0.1%) combined. Its frequency is close to that of SCA3 (2%) and FRDA (2.2%) which are well-established emerging ataxia disorders in India [10].
Amongst the SCA27B positive patients, 21.7% (n = 5) of the patients carry a biallelic expansion between 262 and 645 repeating units, and 8.7% (n = 2) of the patients had a homozygous repeat expansion. The notable occurrence of extensive biallelic expansion in the Indian population is unprecedented in comparison to other populations. This trend could be attributed to the prevalence of consanguineous marriages and endogamy. It is already well established in other polyglutamine SCAs that the severity of the phenotype and the age of onset of a disorder tends to increase with the size of the expansion [26]. In SCA27B, the age of onset is weakly correlated to the repeat length but patients with biallelic expansion may manifest an early onset (below 30 years of age) with or without severe disease phenotype [11, 27]. We had some intriguing observations in a similar vein. Majority of individuals carrying a biallelic expansion exhibited an earlier onset of symptoms. The biallelic expansion may be linked to a change in the disorder’s nature, wherein the haploinsufficiency is altered by a loss-of-function, rendering it more pathogenic. Interestingly, a subset of patients with the short allele repeats in the premutable range also experienced an early onset of symptoms. This implies a potential role of premutable normal alleles and raises the prospect of the existence of associated genetic modifiers.
Among the positive patients with clinical data, 80% of individuals indicated a de novo expansion while 20% of individuals reported a relevant family history in one of the parents indicating the autosomal dominant nature of the disorder. This stochastic de novo manifestation of the disease could be attributed to several factors. Firstly, the FGF14 repeat region is highly variable, ranging between 6 and 223 repeating units in the control cohort, which is in stark contrast to other intronic repeat expansion disorders like FRDA, repeats typically range between 5 and 33 repeating units. This highlights the extreme instability of the FGF14 repeat region. Secondly, previous de novo occurrences of a trinucleotide repeat expansion disorder have been ascribed to “disease anticipation”. SCA27B shows considerable intergenerational instability with maternal anticipation concordance with other trinucleotide repeat expansion disorders such as SCA3 [28], DRPLA [29], and DM1 [30]. Thirdly, incomplete penetrance and phenotypic heterogeneity are usual in autosomal dominant disorders like SCAs having a very narrow range of intermediate repeats [31]. SCA27B, in paradox, has an extensive intermediate repeat ranging between 80 and 250 repeats, however, there’s no clear gap between the pathogenic and non-pathogenic repeat thresholds making it vulnerable to phenotypic heterogeneity and incomplete penetrance over generations. In summary, the de novo expression of the SCA27B phenotype is likely due to the volatile nature of the FGF14 repeat region, intergenerational instability, potential maternal anticipation, and the complex inheritance patterns seen in similar genetic disorders.
The intricate hereditary nature of the disorder led to the investigation of the genomic landscape encompassing the repeat locus. This study reveals a risk haplotype in LD flanking the repeat expansion. The IndiGen dataset (~ 1000 samples) helped us understand the prevalence of the risk haplotype in normal alleles compared to that in expanded and intermediate alleles. The high prevalence of Haplo-1 (AATCCGTGG) in the expanded and intermediate allele highlights it as a prominent risk haplotype in the population. This risk haplotype is in LD across a 74 kb LD block in the Indian population. While the risk haplotype is in LD with normal alleles at the corresponding repeat locus, it indicates a multi-step evolutionary process. This process involves an initial historical mutation giving rise to a large normal allele (proto-mutation). Subsequently, this proto-mutation serves as a reservoir, facilitating gradual expansions that ultimately lead to the development of pathological alleles [26]. The continuous distribution of repeats in SCA27B with no distinct gap between the pathogenic and the non-pathogenic threshold may account for the gradual expansion of the proto-mutant allele. Individuals with the risk haplotype in their proto-mutant allele are at a higher risk of repeat instability during gametogenesis and passing an expanded allele to their posterity.
We further studied the LD from in other populations stipulating a similar 74 kb long stable LD block in the South-Asian population while the other populations (East-Asian, American, African) exemplified partial LD decay. However, the European population lacks a prominent LD region revealing a state of equilibrium. The identified risk haplotype in this region is also highly prevalent in the European population followed by Indian, South-Asian, and American populations. This characteristic makes it more vulnerable to recombination, repeat instability, and expansion. Many of the cohort studies from the European population indicate a high prevalence of the disorder in the population including German (frequency: 8.7–18%) [2, 11], French Canadian (59–61%) [11], French (17%) [32], Greek (12%) [33] and Spanish (28%) [34] cohorts. On the other hand, South-Asian and East-Asian populations have a lower prevalence of the disorder as observed in our Indian cohort (frequency: 1.83%) and Japanese cohort (frequency: 1.2%) [35].
We further studied the age of the mutation in the Indian subcontinent. We followed a correlated genealogy (“tree-like genealogy”) approach as we expected the samples to share a common ancestor earlier than the most recent common ancestor. Moreover, correlated genealogy helps calculate the age of the mutation directly from the data rather than any genealogy model giving genotype-based veritable results. The model also avoids biases by removing any excess sharing between samples with more recent common ancestry than the entire cohort [25]. The study highlights that the repeat expansion mutation is ~ 22,000 years (considering a 20-year generation gap) old suggesting that the most recent common ancestor lived in the Upper Paleolithic age. Considering India’s cultural and socio-economic history, a 20-year generation gap is suitable as early marriages were more prevalent in India. The ancient origin of this mutation even predates Indo-European divergence. Further, we speculate that given the ancient origin of the disorder and its high frequency of mutable GAA-alleles and at-risk haplotypes, may contribute towards the increase in the occurrence of SCA27B in the Indian subcontinent.
Conclusion
The novelty of this study stems from its substantial cohort size, offering legitimate insights into the prevalence of SCA27B in the Indian subcontinent. This study highlights the necessity to screen the genetically unsolved ataxia cases for SCA27B as its prevalence (1.83%) is relevant to that of other emerging ataxia disorders in India. We also identified a potential risk haplotype in linkage disequilibrium providing insights into historical recombination and genealogical relationships among populations, contributing towards a comprehensive understanding of the origin and evolution of the disorder. Further investigations on the association of the at-risk haplotype with the repeat expansion across populations will shed light on the evolutionary trajectory of SCA27B.
Limitations
We acknowledge that while our haplotype analysis replies on SNPs, utilizing microsatellites in studies might yield divergent findings.
Data availability
Data is available to be shared upon request.
References
van Prooije T, Ibrahim NM, Azmin S, van de Warrenburg B (2021) Spinocerebellar ataxias in Asia: prevalence, phenotypes and management. Parkinsonism Relat Disord 92:112–118
Rafehi H, Read J, Szmulewicz DJ, Davies KC, Snell P, Fearnley LG et al (2023) An intronic GAA repeat expansion in FGF14 causes the autosomal-dominant adult-onset ataxia SCA50/ATX-FGF14. Am J Hum Genet 110(1):105–119
Krejci P, Prochazkova J, Bryja V, Kozubik A, Wilcox WR (2009) Molecular pathology of the fibroblast growth factor family. Hum Mutat 30(9):1245–1255
Brusse E, de Koning I, Maat-Kievit A, Oostra BA, Heutink P, van Swieten JC (2006) Spinocerebellar ataxia associated with a mutation in the fibroblast growth factor 14 gene (SCA27): a new phenotype. Mov Disord off J Mov Disord Soc 21(3):396–401
Miura S, Kosaka K, Fujioka R, Uchiyama Y, Shimojo T, Morikawa T et al (2019) Spinocerebellar ataxia 27 with a novel nonsense variant (Lys177X) in FGF14. Eur J Med Genet 62(3):172–176
Coebergh JA, van de Fransen DE, Snoeck IN, Ruivenkamp C, van Haeringen A, Smit LM (2014) A new variable phenotype in spinocerebellar ataxia 27 (SCA 27) caused by a deletion in the FGF14 gene. Eur J Paediatr Neurol EJPN off J Eur Paediatr Neurol Soc 18(3):413–415
Misceo D, Fannemel M, Barøy T, Roberto R, Tvedt B, Jaeger T et al (2009) SCA27 caused by a chromosome translocation: further delineation of the phenotype. Neurogenetics 10(4):371–374
Groth CL, Berman BD (2018) Spinocerebellar Ataxia 27: a review and characterization of an evolving phenotype. Tremor Hyperkinetic Mov 8:534
Brusse E, de Koning I, Maat-Kievit A, Oostra BA, Heutink P, van Swieten JC (2006) Spinocerebellar ataxia associated with a mutation in the fibroblast growth factor 14 gene (SCA27): a new phenotype. Mov Disord 21(3):396–401
Sharma P, Sonakar AK, Tyagi N, Suroliya V, Kumar M, Kutum R et al (2022) Genetics of Ataxias in Indian Population: a collative insight from a Common Genetic Screening Tool. Adv Genet 3(2):2100078
Deep Intronic FGF14 GAA Repeat Expansion in Late-Onset Cerebellar Ataxia (2023) N Engl J Med 388(21):e70
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16(3):1215
Uppili B, Sharma P, Ahmad I, Sahni S, Asokachandran V, Nagaraja AB et al (2023) Sequencing through hyperexpanded Friedreich’s ataxia-GAA repeats by nanopore technology: implications in genotype–phenotype correlation. Brain Commun 5(2):fcad020
Li H (2018) Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34(18):3094–3100
Giesselmann P, Brändl B, Raimondeau E, Bowen R, Rohrandt C, Tandon R et al (2019) Analysis of short tandem repeat expansions and their methylation state with nanopore sequencing. Nat Biotechnol 37(12):1478–1481
Raftery LSC, Fraley T, Brendan Murphy AE (2023) Model-based clustering, classification, and density estimation using mclust in R. Chapman and Hall/CRC, New York, p 268
Rausch T, Hsi-Yang Fritz M, Korbel JO, Benes V (2019) Alfred: interactive multi-sample BAM alignment statistics, feature counting and feature annotation for long- and short-read sequencing. Bioinforma Oxf Engl 35(14):2489–2491
Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27(2):573–580
Dolzhenko E, Deshpande V, Schlesinger F, Krusche P, Petrovski R, Chen S et al (2019) ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics 35(22):4754–4756
Jain A, Bhoyar RC, Pandhare K, Mishra A, Sharma D, Imran M et al (2021) IndiGenomes: a comprehensive resource of genetic variants from over 1000 Indian genomes. Nucleic Acids Res 49(D1):D1225–D1232
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D et al (2007) PLINK: a Tool Set for whole-genome Association and Population-based linkage analyses. Am J Hum Genet 81(3):559–575
Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68(4):978–989
popart full-feature software for haplotype network construction - Leigh – 2015 - Methods in Ecology and Evolution - Wiley Online Library [Internet]. [cited 2024 Mar 2]. https://besjournals.onlinelibrary.wiley.com/doi/full/https://doi.org/10.1111/2041-210X.12410
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21(2):263–265
Gandolfo LC, Bahlo M, Speed TP (2014) Dating rare mutations from small samples with dense marker data. Genetics 197(4):1315–1327
Depienne C, Mandel JL (2021) 30 years of repeat expansion disorders: what have we learned and what are the remaining challenges? Am J Hum Genet 108(5):764–785
Pellerin D, Danzi MC, Renaud M, Houlden H, Synofzik M, Zuchner S et al (2024) Spinocerebellar ataxia 27B: a novel, frequent and potentially treatable ataxia. Clin Transl Med 14(1):e1504
Takiyama Y, Shimazaki H, Morita M, Soutome M, Sakoe K, Esumi E et al (1998) Maternal anticipation in Machado-Joseph disease (MJD): some maternal factors independent of the number of CAG repeat units may play a role in genetic anticipation in a Japanese MJD family. J Neurol Sci 155(2):141–145
Aoki M, Abe K, Kameya T, Watanabe M, Itoyama Y (1994) Maternal anticipation of DRPLA. Hum Mol Genet 3(7):1197–1198
Ho G, Cardamone M, Farrar M (2015) Congenital and childhood myotonic dystrophy: current aspects of disease and future directions. World J Clin Pediatr 4(4):66–80
Paulson H (2018) Repeat expansion diseases. Handb Clin Neurol 147:105
Bonnet C, Pellerin D, Roth V, Clément G, Wandzel M, Lambert L et al (2023) Optimized testing strategy for the diagnosis of GAA-FGF14 ataxia/spinocerebellar ataxia 27B. Sci Rep 13(1):9737
Kartanou C, Mitrousias A, Pellerin D, Kontogeorgiou Z, Iruzubieta P, Dicaire MJ et al (2024) The FGF14 GAA repeat expansion in Greek patients with late-onset cerebellar ataxia and an overview of the SCA27B phenotype across populations. Clin Genet
Iruzubieta P, Pellerin D, Bergareche A, Albajar I, Mondragón E, Vinagre A et al (2023) Frequency and phenotypic spectrum of spinocerebellar ataxia 27B and other genetic ataxias in a Spanish cohort of late-onset cerebellar ataxia. Eur J Neurol 30(12):3828–3833
Ando M, Higuchi Y, Yuan J, Yoshimura A, Kojima F, Yamanishi Y et al (2023) Clinical variability associated with intronic FGF14 GAA repeat expansion in Japan. Ann Clin Transl Neurol 11(1):96–104
Acknowledgements
We are grateful to all the participants and family members for enrolling in the study.
Funding
This work was supported by ICMR 5/4–5/5Ad-hoc/Neuro/220/NCD-I (GAP240). We also acknowledge the ICMR-SRF (Project:2021–8342/Genomics-BMS) fellowship of Ms. Pooja Sharma.
Author information
Authors and Affiliations
Contributions
T.D and P.S: Performed all the experiments, conducted the analysis and Manuscript writing; B.U.: performed Haplotype analysis; V.A.: Long read sequencing data analysis; S.B: conducted age of mutation analysis; AKS-1: Clinical data analysis; AKS: Clinical Data analysis and reviewing of the work; M.F.: Conceptualization, Design and manuscript writing.
Corresponding author
Ethics declarations
Compliance with ethical standards
Institutional Human Ethics Committees (GAP-240) of the participating institutes have approved this study. All the participants had given their informed consent for the study.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
De, T., Sharma, P., Upilli, B. et al. Spinocerebellar ataxia type 27B (SCA27B) in India: insights from a large cohort study suggest ancient origin. Neurogenetics (2024). https://doi.org/10.1007/s10048-024-00770-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10048-024-00770-y