
Abstract
A PdAg deposit containing ~ 25 at.% Ag is obtained by the electrochemical codeposition from an aqueous solution of Pd and Ag sulfates (Au support, 0.5 M H2SO4). The deposit is characterized by means of various physical, physicochemical, and electrochemical methods. The PdAg deposit demonstrates the ~ 2 times higher specific activity (per the electrochemically active surface area (EASA) of Pd) in the formic acid oxidation reaction (FAOR) as compared with the individual Pd deposit prepared under the same conditions. The effect of silver additions on the palladium activity depends on many factors. The corrosion stability of PdAg is studied in 0.5 M H2SO4 solution based on the overall cyclic voltammograms (CVAs) and also on anodic and cathodic half-cycles in the region E = 0.3 − 1.25 V (vs. reversible hydrogen electrode (RHE)). The electrochemical estimates are compared with the results of direct analytical determination of dissolution products in solution after anodic polarization of deposits. The total amounts of Pd dissolved substantially increase with incorporation of Ag, which is associated, first of all, with the considerable increase in the EASA; at the same time, the specific dissolution of Pd also substantially increases. The possible factors determining the active dissolution of PdAg deposits are discussed; in particular, the specific mechanism of their dissolution via silver adatoms is proposed.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The Pd–Ag composites are widely studied because they are cheaper than pure palladium catalysts, and their activity in various reactions is often higher as compared with palladium. Among the latter reactions, mention should be made of the formic acid oxidation reaction (FAOR) [1,2,3,4,5,6,7,8], the oxygen reduction reaction (ORR) [3, 9, 10], the synthesis of hydrogen by catalytic decomposition of HCOOH [11,12,13], and dehalogenation [14, 15]. For longer than 50 years, attention was focused on the FAOR because formic acid serves as the fuel in low-temperature fuel cells with proton-exchange membranes (PEMFC). Being the best monometallic catalyst for the formic acid oxidation [16, 17], palladium fails nonetheless to provide the HCOOH oxidation rates sufficient for the commercial use of PEMFC at room temperature. It was found [3, 18,19,20,21,22,23,24] that the Pd activity in FAOR can be substantially enhanced by the addition of certain inexpensive d-metals (Cu, Pb, Co, etc.). The literature data on the effect of silver inclusions to palladium on its FAOR activity are very controversial. Thus, in certain investigations, the transition from palladium to Pd–Ag composites was observed to enhance the catalytic activity [1,2,3,4, 20]; in the other studies, no noticeable catalytic effects were observed [5, 6]. In [3, 4], the FAOR activity of Pd–Ag catalysts was found to depen strongly on the way these two components were mixed and also of the Ag:Pd ratio.
A serious obstacle to the use of palladium as the catalyst is its low corrosion stability, the lowest among platinum-group metals [18, 22, 25,26,27,28,29,30,31,32,33,34,35,36,37,38]. The problem of palladium dissolution was considered in numerous studies; however, the majority of fundamental investigations were carried out for compact palladium samples [18, 25,26,27,28,29,30,31]. The kinetics and mechanism of electrodissolution of disperse palladium were studied in less detail [32,33,34,35,36]. Moreover, the question of how the addition of the other metals affects the corrosion stability of disperse palladium still remains open [18, 22, 37,38,39,40]. From the viewpoint of the use of palladium-based catalysts in fuel cells with proton-exchange membranes, the studies of palladium electrodissolution in sulfuric acid solutions at potentials below 1.23 V (RHE) deserve the keenest attention. In several studies [4, 22, 37, 39], the incorporation of foreign metals was observed to accelerate the palladium dissolution; however, there are also studies [18, 33, 38, 40] in which the corrosion stability of PdM was found to be higher as compared with Pd. The comparative analysis of these results is extremely complicated due to the great difference in the degrees of dispersion of PdM alloys, the Pd:M ratios, the electrode polarization conditions, etc. Conclusions on the corrosion stability of bimetallic catalysts are often made based on indirect factors such as the changes in the true surface area and/or specific mass activity at accelerated stress-testing or acidic treatment in the O2 atmosphere [18, 37, 38, 41, 42]. Moreover, the effects of palladium dissolution may be compensated by changes in the degree of dispersion and/or specific surface activity. The question whether palladium dissolves mainly in the course of its anodic polarization (in the anodic scan) or during the cathodic reduction of oxides is still debated [25,26,27,28, 30,31,32, 43, 44].
In our previous studies [22, 40, 44], to compare the electrodissolution of PdM alloys and individual Pd, we studied their electrodeposits (ED). The electrodeposition allows obtaining sufficiently dispersed structures without using any surface-active organic stabilizers. For the quantitative determination of the dissolution of samples, we have proposed the method of measuring the anodic and cathodic “half-cycles” in the interval E = 0.3–1.25 V, which was followed by analysis of dissolution products in solution. The use of this method made it possible to separate more precisely the dissolution in the cathodic and the anodic potential scans and elucidate the role of oxides in the electrodissolution of the deposit. The total amount of Pd dissolved from the PdPb and PdCu deposits was found to be much higher as compared with individual Pd deposits, which was associated mainly with the substantial increase in the degree of dispersion of deposits upon inclusion of relatively low amounts of Pb or Cu. At the same time, we have noticed that in the presence of copper additive, the specific dissolution of Pd was substantially lower. Comparing the dissolution of deposits in the anodic and cathodic half-cycles allowed us to conclude that the dissolution proceeds preferentially by the direct reaction Pd → Pd2+ and that oxides hinder the dissolution process. The inhibiting effect of oxides on the dissolution of the mixed deposit PdMo was also observed in [45]. In [4], the dissolution of Pd from its mixed deposit Pd0(Ag) prepared by galvanic displacement was observed to be higher as compared with the individual Pd deposit. It seemed interesting to carry out the more systematic investigations of the effect of small additions of silver on the dissolution of palladium using the method described in [22, 40]. In this study, we studied the formic acid oxidation only in the low-potential region where the dissolution of both Pd and Ag was absent. However, the results on the electrodissolution of PdAg in the potential region of oxygen adsorption can also be important for the storage of anodic catalysts and also for the determination of their stability at short circuiting in direct formic acid fuel cells (accidental or aimed at periodic removal of strong chemisorption products that can inhibit the FAOR).
Experimental
All electrochemical measurements were carried out in a three-electrode cell with separate anodic and cathodic compartments in the argon atmosphere at 20 ± 1 °C. Potentiostates R301M (“Elins”) and P-5827 (Russia) were used. For preparation of solutions, we used H2SO4 (Merck Suprapur), Ag2SO4 (Fluka, puriss p.a.), PdSO4 (Aldrich), HCOOH (Fluka puriss p.a.), and water purified in the Milli-Q system.
Pd and Ag were codeposited onto polycrystalline Au plates (0.8 cm2 on both sides) under potentiostatic conditions from solution of 10−3 M PdSO4 + × 10−4 M Ag2SO4 + 0.5 M H2SO4 (pH 0.35) at Edep = 0.4 V (from here on, the potential values E are related to the reversible hydrogen electrode in 0.5 M H2SO4 solution). The 1.1 C charge was passed, which corresponded to the deposition of ~ 0.6 mg Pd from a solution that contained no silver salts (deposit thickness ~ 0.5 μm). The current efficiency of metal deposition was ~ 90%. From solutions with x = 1, the electrodeposits (ED) containing 24 ± 3 at.% Ag were obtained. These deposits were chosen for the further studies, because in regard the content of the non-noble component they were close to the PdCu and PdPb deposits used earlier in corrosion tests [22, 40]. For the EDs PdAg with the high Ag content (≥ 50 at.%), no increase in the FAOR activity was observed as compared with ED Pd [6]; the promoting effect is expected at the lower silver content. The Pd electrodeposits used for a comparison were deposited from the 10−3 M PdSO4 + 0.5 M H2SO4 solution under the same conditions as the PdAg deposit. The FAOR activity of deposits was tested based on the value of the stationary current measured under potentiostatic conditions in the solution of 0.5 M H2SO4 + 0.5 M HCOOH (pH ~ 0.3); the stationarity criterion corresponded to variations of the current < 2% per min.
The SEM images of deposits were obtained using the device JEOL-JSM6490 LV at an accelerating voltage of 30 kV. The bulk composition and the structure of deposits were determined by the methods of energy-dispersive X-ray spectroscopy (EDX), X-ray diffraction analysis (XRD), and inductively coupled plasma atomic emission spectroscopy (ICP-AES). Their surface composition was determined using X-ray photoelectron spectroscopy (XPS); the XPS spectra were collected by means of a spectrometer Axis Ultra DLD, Kratos (monochromic AlKα radiation with the analyzer energy of 160 eV). Bruker D8 ADVANCE diffractometer with CoKα radiation (wavelength = 0.179026 nm) was used for studying the lattice parameter of deposits.
The electrochemically active surface area (EASA) of samples was determined from the charge consumed in the desorption of a monolayer of Oads in the cathodic scan of cyclic voltammogram (the normalization factor was 420 µC cm−2 [46, 47]). We had to abandon the idea to determine the EASA based on copper adatoms because we observed considerable incorporation of copper into the surface layer of deposits, which could substantially affect their electrocatalytic behavior.
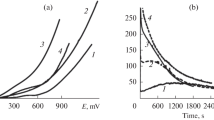
The dissolution of deposits was carried out in deaerated solutions of 0.5 M H2SO4 at the linear potential scanning with a rate v = 2.0 mVs−1. Figure 1 shows five dissolution modes. The modes IIan and IIcat are called the anodic and the cathodic half-cycles, respectively. The use of the anodic half-cycles allowed us to eliminate the dissolution and the redeposition of Pd in the cathodic scan that occur when the full-cycle voltammogram (CVA) is measured, whereas the use of the cathodic half-cycles made it possible to eliminate the dissolution in the anodic scan [22, 44]. The concentration of metals in solution was determined by the ICP-AES technique. It deserves mention that the corrosion mode I and also modes II and III were used for freshly prepared deposits. At the same time, before starting the studies on the electrodissolution of deposits, we measured considerably fast full CVAs (v = 20 mV s−1) in 0.5 M H2SO4 in the interval E = 0.3 − 1.25 V in order to determine the EASA and also for certain stabilization of the deposit surface. The slow CVAs were measured in a new portion of deaerated solutions of 0.5 M H2SO4.

Scheme of potential variation in the experiments on the comparison of dissolution of PdAg and Pd deposits during anodic polarization (see text)
Results and discussion
Morphology and composition of deposits
Figure 2c shows that the palladium deposits consist mainly of lamels between which the inclusions of individual spherical globules of 0.4–0.7 µm are located. The electrochemical codeposition of Pd and Ag led to the formation of the smaller “loose globules” of 0.15–0.5 µm which were distributed sufficiently uniformly over the gold surface (Fig. 2a). The SEM images make it possible to assume that the degree of dispersion of the Pd deposit increases after the incorporation of small amounts of silver. However, this assumption should be confirmed by some other method because the globules observed by SEM may turn out to be agglomerates of finer particles (see the next section). The slow potential cycling (mode I) made the Pd deposit more homogeneous; however, the size of globules remained unchanged (Fig. 2d). The morphology of the PdAg deposit after its polarization in mode I remained virtually unchanged (Fig. 2b). The changes in SEM images for deposits cycled in mode II were also minor. Such an effect of cycling suggests that the deposit layers removed in these cycles were very thin as compared with the size of particles observed by SEM.

SEM images of (a, c) freshly prepared (a) PdAg and (c) Pd deposits and (b, d) after their polarization in mode I: (b) PdAg and (d) Pd
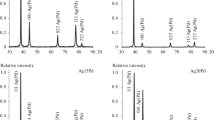
The EDX spectra of the PdAg deposits after the experiments on their dissolution turned out to be close to one another and also to the spectrum of the untreated mixed deposits (for example, Fig. 3 shows the EDX spectra for two samples). The estimated bulk content of silver (Pd + Ag = 100 at.%) also showed the absence of considerable changes in the bulk composition after the electrodissolution in the modes used (Table 1). A small decrease in the bulk content of silver was observed for the sample polarized in mode I. This pointed to the predominant dissolution of Ag from the two-component PdAg deposit. On the whole, the EDX data provided the additional evidence for the dissolution of small amounts of deposits.

EDX spectra of (a) freshly prepared PdAg deposit and (b) after its polarization in mode IIan
Figure 4 shows the XRD patterns of deposits. The lower pattern belongs to an Au plate with the Pd deposit. Because the Pd layer was thin, the gold peaks were quite intense. The position of gold peaks well coincided with the reference data [48]. The lattice parameter for the Pd deposit is 0.3892 nm (the reference data for Pd is 0.3890 nm [49]). In the PdAg deposit, we observed the presence of the Pd phase and also of a new disordered phase of PdxAg(1−x) [50]. After the electrodeposition, the peaks of Pd slightly shifted to the lower angles, which corresponded to the increase in the lattice parameter to 0.3906 nm. The lattice parameter of this newly formed phase of PdxAg(1−x) is 0.3981 nm. According to the previous studies, this new phase is Pd0.49Ag0.51 [49, 50]. The regions scanned by EDX are several tens of micrometers; for XRD, the size of inspected regions is several millimeters. Insofar as the size of particles in the PdAg deposit is much smaller, the results obtained by EDX and XRD give no way of concluding anything on the location of these two phases in the mixed deposit.

XRD pattern for Pd (lower line) and PdAg (upper line) deposits
Figure 5 shows the overview XPS spectra for several samples of ED PdAg. Table 1 shows the content of palladium and silver (Pd + Ag = 100 at.%) on samples’ surface calculated based on the XPS data. It is seen that after the freshly deposited PdAg sample was polarized anodically in different modes, a large amount of Ag remained on its surface. Only after its polarization in mode IIcat, a substantial decrease in the surface content of Ag as compared with initial ED PdAg was observed. It is interesting that a slight decrease in the Ag content in the surface layer was also noted in [4] during potential cycling (0.09–1.4 V) for the Pd0(Ag) composite prepared by galvanic displacement. Presumably, this was mainly associated with the presence of silver adatoms on Pd at the potentials of oxygen adsorption [4] and the fast transition PdAg → PdAgad. It should be noted that the content of alloy components in the surface layer is variable because when the samples are polarized under the chosen conditions, considerable amounts of both Ag and Pd pass to solution (see the final section).

Overview XPS spectra of (1) freshly prepared PdAg deposits, after their polarization in modes (2) I, (3) IIan, (4) IIcat; (5) e.d. Pd
The analysis of silver and palladium lines in the XPS spectra and also of the Auger spectra of these elements (are not shown here due to their similarity to the analogous spectra of Pd0(Ag) [4]) has shown that both metals are present on the surface preferentially in their nonoxidized state. The Ag3d XPS spectra of samples showed the doublet of slightly asymmetrical narrow lines of the Ag3d5/2 component with the binding energy of 367.5 eV, which is lower by approximately 0.7 eV as compared with pure metal silver. Such a shift is typical of Pd–Ag alloys with the silver content below 30 at.% [51].
Voltammograms of deposits
Figure 5 shows the typical CVAs for PdAg (a,b) and Pd (c,d) deposits measured at the fast (a,c) and slow (b,d, mode I) potential scan rates. Curves a and c which were measured for estimation of the EASA and also for the surface stabilization (see “Experimental”) substantially differ even visually in the potential region of oxygen adsorption (> 600 mV), which is associated with the increase in both the EASA and the dissolution of the Pd deposit upon the inclusion of relatively low amounts of silver. The integral anodic charge at E > 600 mV (the positive currents were summarized in both anodic and cathodic scans) minus the corresponding integral cathodic charge at v = 20 mVs−1 was found to be equal to ~ 0.7 mC for Pd and ~ 60 mC for PdAg. This pointed to the strong increase in the deposit dissolution in the presence of silver (for PdAg, this charge corresponded to the sum of dissolution charges of Pd and Ag) [25].
Attention should be drawn to the splitting of the cathodic peak to two peaks in the first fast CVA recorded on the freshly prepared mixed deposit (Fig. 6a). Based on the XRD data, the latter effect can be associated with the exposure of two phases to the surface. However, it should be taken into account that the XRD data characterize the bulk structure of the deposit, whereas the CVA describes the processes on the surface, i.e., in a very thin layer which cannot be analyzed even by XPS (XPS analysis depth ~ 3 nm). It is evident that immediately after its deposition, the surface layer of the alloy is in the extremely unstable state. In the second and third cycles of fast CVAs (e.g., Fig. 7) and also in the first slow CVA (Fig. 6b), the splitting of the cathodic peak was absent. Apparently, the restructuring of the surface layer was very fast and led to its higher degree of homogeneity. We can also assume that in case of the separate location of two phases in the freshly deposited PdAg, the splitting of the cathodic peak (Fig. 6a) would not disappear so soon during electrode potential cycling (Figs. 8 and 9).

CVA of freshly prepared deposits of (a, b) PdAg and (c, d) Pd measured in 0.5 M H2SO4 at v, mV s.−1: (a, c) 20 and (b, d) 2

CVA of ED PdAg in solution of 0.02 M CuSO4 + 0.5 M H2SO4. Inset: CVA of ED Ag in solution of 0.5 M H2SO4 (see text)

Stationary polarization curves of FAOR in 0.5 M HCOOH + 0.5 M H2SO4 on (1) ED PdAg (23 at.% Ag, XPS), (2) ED Pd, and (3) Pd0(Ag) deposit obtained by galvanic displacement (28 at.% Ag, XPS) [20]. The current is normalized to EASA

(a) Anodic and (b) cathodic half-cycles in mode II (see text)
The assessment of the EASA of Pd from the charge consumed in the oxygen electrodesorption gave 5 ± 1 cm2 for Pd deposits and 30 ± 8 cm2 for PdAg (the data for a large number of deposits used in this study were averaged). It is evident that the increase in EASA should be considered among the main factors responsible for the increase in the overall dissolution of palladium when silver is present in the deposit [22, 40]. Assessing the average size of particles based on the EASA by the model of spheres gave the value 0.56 ± 0.1 µm for Pd, in good agreement with the SEM data, which points to the low internal porosity of the deposit. The similar assessment of the particle size in the PdAg deposit (with regard to the fact that palladium occupied ~ 75% of the overall surface) gave the value 0.11 ± 0.03 µm. This confirmed the considerable increase in the degree of dispersion of the deposit as a result of incorporation of silver. At the same time, the latter value was considerably lower than the particle size in the PdAg deposit assessed based on the SEM data. This difference can be associated with the high surface roughness of globules and/or certain conglomeration of particles.
An attempt was undertaken to assess the possible involvement of silver into the process of oxygen adsorption on PdAg by the comparison with the CVA measured on an individual silver deposit. However, this turned out to be impossible because the adsorption of oxygen on PdAg was accompanied by the high currents of silver dissolution and also due to the presence of cathodic current of the electroreduction of the passivating film of nonsoluble silver sulfate deposit (insert in Fig. 7). The potential of cathodic current maximum is close to the standard potential of the half-reaction Ag2SO4 + 2e = 2Ag + SO4−2− (0.653 V).
As the potential scan rate decreased, the shape of CVAs in the anodic potential region for Pd and PdAg (Fig. 5b and d) underwent no substantial changes. The integration of the current at v = 2 mVs−1 demonstrated a considerable increase in the dissolution of the Pd deposit (by a factor of ~ 5 according to the charge consumed in electrodissolution), whereas the dissolution of PdAg remained at the same level (in the charge units) as v decreased tenfold. The behavior of PdAg seems to be anomalous and allows one to assume that the structure of the freshly prepared mixed deposit is more defective as compared with the structure of the analogous deposits of individual Pd.
It is well known that the potential of the cathodic peak of oxygen electrodesorption characterizes the binding energy of adsorbed oxygen. According to Fig. 5, this potential for the Pd deposit is 750 mV at v = 20 mVs−1 and 790 mV at v = 2 mVs−1; for PdAg, this potential is 700 (the average value for two peaks) and 740 mV, respectively. First, the considerable difference of the potentials of Oads desorption for PdAg and Pd agrees with the above assumption on the presence of a bimetallic phase (or phases) on the surface of the freshly prepared PdAg deposit. Second, the decrease in the cathodic peak potential (at the same v) when going from Pd to PdAg suggests that the bond formed by chemisorbed oxygen with palladium strengthens upon introduction of silver.
Formic acid oxidation
We restricted our consideration to the stationary FAOR currents in the low-potential region (< 300 mV) because the latter region is very interesting from the standpoint of using formic acid in fuel cells. Figure 6 shows that on average, the introduction of silver in small amounts to the palladium deposit doubles its specific activity in FAOR (curves 1 and 2). The authors of several studies [1,2,3,4] observed the stronger activation effect of the silver molecules introduced to Pd on the oxidation rate of formic acid in acidic solutions. At the same time, in [6], the absence of any noticeable effect was observed at incorporation of ~ 50 at.% Ag into the mixed ED PdAg.
We can assume that this discrepancy is associated, first of all, with the different contents of silver in the surface layer of composites. To check this assumption, we compared the specific FAOR rates for several Pd–Ag structures considered in [4, 6, 20] and in this study. The results of these studies were chosen because their authors also used the disperse deposits formed by electrodeposition from solutions containing neither structure-forming surface-active organic compounds nor chloride ions. Furthermore, the electrochemical methods used for determination of EASA were the same, the FA electrooxidation was studied in one and the same solution, and the same criterion of stationarity current was used. All these factors were required for the correct comparison of the results. Table 2 shows the stationary FAOR currents at 200 mV on the PdAg deposits prepared by the codeposition (samples 1 and 2) and the galvanic displacement of ED Ag by palladium (Pd0Ag, samples 4–6) with indication of the fraction of silver contained in the surface layer of Pd0Ag composites (XPS data). Figure 6 gives, as an example, one of polarization curves of FAOR on Pd0Ag (sample 3 in Table 2). Although curves 2 and 3 were measured on samples with the close contents of Ag in their surface layer (23 and 28 at.%, respectively), their current differed ~ fourfold. Table 2 shows the absence of any correlation between the surface layer composition and the activity of PdAg in FAOR. Indeed, it should be taken into account that the XPS method determines only the average composition of a ~ 3-nm layer, which corresponds to more than 10 monolayers. However, the absence of the expected correlation once again confirms that the effect of the addition of the second component (M) to the Pd catalyst depends on many factors. These factors may include the size effect, the third body effect, the electronic effect, the effect of lattice defects, the formation of new active sites (ensembles), and the structure of the Pd/M interface [1,2,3,4,5,6]. It is especially difficult to single out the dominating factor(s) for the catalysts with the highly developed surface.
Electrodissolution of the PdAg deposit
In the section “Voltammograms of deposits,” certain preliminary results were shown on the dissolution of PdAg and Pd deposits. These results were obtained exclusively by electrochemical measurements. To gain deeper insight into the effect of small silver inclusions to palladium on the deposit dissolution, we used the direct analytical measurements of the metals that had passed to solution during the anodic polarization of deposits.
Table 3 compares the data on the dissolution of ED PdAg in five modes of anodic polarization (Fig. 1) with the analogous data for individual ED Pd deposited also at 0.4 V. Here, m is the total amount of metal passed to solution; ms(PdAg) and ms(Pd) are the values of m(Pd) for the corresponding deposits normalized to the average EASA. Table 3 shows that the overall dissolution of palladium from PdAg much exceeds the dissolution of the individual Pd deposit. This is mainly associated with the increase in the degree of dispersion of the deposit upon introduction of silver. However, the ratio of m(Pd) values summarized over the modes I, IIan, IIIan, and IIcat for ED PdAg to the analogous sum for ED Pd is ~ 9 and substantially exceeds the ratio between the average EASA values of mixed and individual palladium deposits (~ 6). The considerable increase in the specific dissolution of the deposit after the incorporation of silver is also confirmed by the ratio of nonintegrated values ms(PdAg)/ms(Pd) (Table 3) which exceeds 1 by a factor of ~ 1.5–3.
Quite unexpected was the absence of any substantial difference between the dissolution of the PdAg deposit in anodic and cathodic half-cycles (Table 3). The earlier studies of ED PdPb and PdCu [44] revealed the much stronger dissolution in the anodic half-cycles as compared with the cathodic half-cycles. This was explained by the inhibiting effect of oxides on the electrodissolution process. To find the reasons for the specific behavior of the PdAg deposit, the analytical data on its dissolution were compared with the electrochemical estimates of its dissolution in half-cycles. By integrating the current in the anodic half-cycle IIan (Fig. 7a) and correcting the result for the electric double-layer charging and the oxygen adsorption (0.420 mC cm−2 × 30 cm2), we obtained the charge of ~ 43 mC which was consumed in the dissolution of palladium and silver. At the same time, under the assumption that 2e is consumed per Pd atom and 1e per Ag atom, the analytical data showed that the charge consumed in mode IIan was equal to ~ 38 mC, which agreed adequately with the electrochemical estimate. The main peculiarity of the cathodic half-cycle (Fig. 7b) is the high anodic current in the potential region of 1.25–0.90 V. This current comprises the currents of the electric double-layer charging, the oxide formation, and the dissolution of alloy’s metals. Based on the conclusion drawn in [30, 44] that palladium dissolves mainly from the surface areas free of oxide, it can be expected that in the case of the fast formation of the PdO monolayer as the potential shifts to 1.25 V, a sharp drop of anodic current should occur in the beginning of the cathodic scan at least as a result of blocking of the Pd dissolution. However, the analytical measurements (Table 3) showed the presence of considerable dissolution of palladium in mode IIcat. This can be explained by the absence of a full PdO monolayer due to its slow formation and/or by the nonmonolayer character of the limiting coverage of the Pd surface by chemisorbed oxygen at 1.25 V. The latter effect was also noted in [35] for several forms of ED Pd. The integration of the current in the cathodic peak at ~ 0.8 V gave the charge corresponding to ~ 0.6 monolayer of Oads. Thus, the above results agree with the assumption that the preferential dissolution of palladium does not proceed via oxide [30, 44].
The amounts of silver m(Ag) dissolved from its codeposit with Pd in all anodic polarization modes exceeded several times the dissolution of the individual Pd deposit (the molar ratio Ag:Pd is 2–5, Table 3). The so strong difference in the dissolution of components of mixed ED PdM was not observed in [22, 40] for M = Pb, Cu.
It is interesting to compare the results found for the PdAg deposit with the data for the PdCu deposit [40]. The latter deposit was synthesized by the same procedure as the PdAg deposit from sulfates of both components at the same deposition potential of 0.4 V; its electrodeposition was also carried out in 0.5 M H2SO4. These factors make the comparison more correct. Table 3 (lower part) also shows the data for PdCu [40]. It is seen that the values of m(Pd) for these two mixed deposits differ insignificantly, whereas the specific dissolution values ms(Pd) for PdCu are much lower as compared with PdAg. The lower stability of PdAg deposits as compared with PdCu can be associated to a certain extent with the higher binding energy between Pd and Cu atoms (the mixing heat Pd-M is 7 kJ/mol [52] for Cu and 5 kJ/mol [53] for Ag) and also with the larger radius of Ag atoms rAg(1.45 nm) > rPd(1.40) > rCu(1.35). For such a relation of radii, the incorporation of silver atoms into the surface layer of Pd should induce tensile strains, whereas the inclusion of Cu atoms should induce compressive strains. The tensile strains decrease the corrosion stability of palladium in its alloys [1]. The relation between the radii of these three metals correlates with the increase in the specific dissolution of Pd upon incorporation of silver and its decrease in the case of copper. A strong increase in the electrodissolution of Pd in perchloric acid solution was observed at the incorporation of Pb into a Pd deposit (rPb = 1.60 nm) [22]. However, it seems unlikely that the relation between radii is the decisive factor. A significant role is also played by the deposit structure and the formation of oxides on its surface [44]. Moreover, the silver adatoms can be involved in the process of dissolution of the PdAg deposit in the potential region under consideration. When the potential of the onset of Pd dissolution is reached (~ 0.7 V), the copper adatoms are already completely removed from the surface, whereas the silver adatoms only begin to leave the Pd surface (their complete removal occurs at E > 1.25 V [54]). The dissolution can proceed by the following scheme: PdAg → PdAgad → Pd2+ + Ag+ + 3e.
The much more active dissolution of silver as compared with copper from their binary alloys with Pd (Table 3) makes it possible to assume the more homogeneous dissolution of PdCu as compared with PdAg. This result is quite unexpected. The standard potential of the pair Ag/Ag+ (E0 = 0.799 V) much exceeds the standard potential of the pair Cu/Cu2+ (E0 = 0.345 V), which allows one to expect the more active copper dissolution. Presumably, the observed effect is associated with the higher energy of the Pd-Cu bond on the surface as compared with the Pd–Ag bond. At the same time, it is possible that the mechanisms of dissolution of copper and silver from their alloys with palladium substantially differ, which is associated with the aforementioned difference in the formation of the adatomic copper and silver coatings on palladium. Silver adatoms can play the active role in the dissolution of silver from the PdAg alloy. Silver dissolution may proceed by the following reaction: PdAg → PdAgad → Pd + Ag+ + e.
Conclusions
-
Mixed deposit PdAg (~ 25 at.% Ag) is obtained by electrodeposition under potentiostatic conditions from the solution of 10−3 M PdSO4 + 10−4 M Ag2SO4 + 0.5 M H2SO4. The deposit is characterized by the SEM, XRD, XPS, ICP-AES, and CVA methods. The formation of solid solution is confirmed. The incorporation of silver leads to the ~ sevenfold increase in the EASA of Pd.
-
The incorporation of silver into Pd increases the specific activity of Pd in FAOR about 2 times. The analysis of this effect which involved the literature data has shown that the changes in the activity upon the addition of silver to Pd deposits do not correlate with the surface composition of codeposits but depend on many factors.
-
By using the ICP-AES, CVA methods, and the method of potentiodynamic half-cycles, the electrodissolution of the PdAg deposit was compared with electrodissolution of the Pd deposit prepared under similar conditions. It is found that the total amounts of Pd dissolved strongly increase after the addition of Ag, which is associated first of all with the increase in the EASA. At the same time, the specific dissolution of Pd also substantially increases.
-
The dissolution of Pd proceeds preferentially on the surface areas free of oxides. It is assumed that the anomalous dissolution of the PdAg deposit and the high Ag:Pd ratio at the electrodissolution are associated with the involvement of silver adatoms in this process.
Change history
17 May 2023
A Correction to this paper has been published: https://doi.org/10.1007/s10008-023-05517-7
References
Jiang K, Zang H-X, Zou Sh, Cai WB (2014) Electrocatalysis of formic acid on palladium and platinum surfaces: from fundamental mechanisms to fuel cell applications. Phys Chem Chem Phys 1516:20360–20376
Chen D, Cui P, Liu H, Yang J (2015) Heterogeneous nanocomposites composed of silver sulfide and hollow structured Pd nanoparticles with enhanced catalytic activity toward formic acid oxidation. Electrochim Acta 153:461–467
Jiang X, Wang Y, Wang J, Li N, Zhou J, Fu G, Sun D, Tang Y (2019) Treelike two-level PdxAgy nanocrystals tailored for bifunctional fuel cell electrocatalysis. J Mater Chem A 7:5248–5257
Podlovchenko BI, Maksimov YuM, Maslakov KI, Volkov DS, Evlashin SA (2017) Galvanic displacement and electrochemical leaching for synthesizing Pd-Ag catalysts highly active in FAOR. J Electroanal Chem 788:217–224
Chen D, Cui P, He H, Liu H, Yang (2014) Highly catalytic hollow palladium nanoparticles derived from silver@silver-palladium core-shell nanostructures for the oxidation of formic acid. J Power Sources 272:152–159
Podlovchenko BI, Maksimov YuM, Utkin AG (2015) Formation of the core–shell structure in the Pd–Ag system by electroleaching of the alloy. Electrocatalytic Properties Russ J Electrochem 51:891–898
Cui Zh, Yang M, DiSalvo FrJ (2014) Mesoporous Ti0.5Cr0.5N supported PdAg nanoalloy as highly active and stable catalysts for the electro-oxidation of formic acid and methanol. ACS Nano 8:6106–6113
Fu G-T, Liu Ch, Zhang Q, Chen Y, Tang Y-W (2015) Polyhedral palladium–silver alloy nanocrystals as highly active and stable electrocatalysts for the formic acid oxidation reaction. Sci Rep 5:Article number 13703
Wang T, Chutia A, Brett Dan JL, Shearing PR, He G, Chai G, Parkin Ivan P (2021) Palladium alloys used as electrocatalysts for the oxygen reduction reaction. Energy Environ Sci 14:2639–2669
Betancourt LE, Arnulfo R-P, Orozco I, Frenkel AI, Li Y, Sasaki K, Senanayake Sanjaya D, Cabrera CR (2020) Enhancing ORR performance of bimetallic PdAg electrocatalysts by designing interactions between Pd and Ag. ACS Appl Energy Mater 3:2342–2349
Tedsree K, Li T, Jones S, Chan ChWA, Yu KMK, Bagot PAJ, Marquis EA, Smith GDW, Tsang ShChEd (2011) Hydrogen production from formic acid decomposition at room temperature using a Ag–Pd core–shell nanocatalyst. Nat Nanotechnol 6:302–307
Mandal K, Bhattacharjee D, Dasgupta S (2015) Synthesis of nanoporous PdAg nanoalloy for hydrogen generation from formic acid at room temperature. Int J Hydrog Energy 40:4786–4793
Yu W-Y, Mullen GM, Flaherty DW, Mullins CB (2021) Selective hydrogen production from dechlorination of 2,4-dichlorophenoxyacetic acid on a roughened silver–palladium cathode. Electrochim Acta 96:90–96
Xu YH, Cai QQ, Ma HX, He Y, Zhang H, Ma CA (2013) Optimisation of electrocatalytic dechlorination of 2,4-dichlorophenoxyacetic acid on a roughened silver–palladium cathode. Electrochim Acta 96:90–96
Brudzisz A, Rajska D, Gajewska M, Sulka GD, Brzózka A (2020) Controlled synthesis and characterization of AgPd nanowire arrays for electrocatalytic applications. J Electroanal Chem 873:114373
Parsons R, VanderNoot T (1988) The oxidation of small organic molecules: a survey of recent fuel cell related research. J Electroanal Chem 257:9–45
Petrii OA (2019) The progress in understanding the mechanisms of methanol and formic acid electrooxidation on platinum group metals (a review). Russ J Electrochem 55:1–33
Meng H, ZengXie DR (2015) FY Recent development of Pd-based electrocatalysts for proton exchange membrane fuel cells. Catalysts 5:1221–1274
Morales-Acosta D, Ledesma-Garcia J, Godinez LA, Rodriguez HC, Alvarez-Contreras L, Arriaga LC (2010) Development of Pd and Pd–Co catalysts supported on multi-walled carbon nanotubes for formic acid oxidation. J Power Sources 195:461–465
Maksimov YuM, Podlovchenko BI, Gallyamov EM, Dagesyan SA, Sen VV, Evlashin SA (2017) Galvanic displacement of silver deposited on carbon nanowalls by palladium and the electrocatalytic behavior of the resulting composite. Mendeleev Commun 27:382–384
Hu Sh, Scudiero L, Ha S (2014) Electronic effect of Pd-transition metal bimetallic surfaces toward formic acid electrochemical oxidation. Electrochem Commun 38:107–109
Podlovchenko BI, Maksimov YuM, Volkov DS, Evlashin SA (2020) Codeposition of Pd and Pb and electrocatalytic properties of their composite. J Electroanal Chem 858:113787
Li R, Yuan H, Ma Z, Tang B, Li J, Wang X (2019) Facile coupling of content design and efficient modulation on the activity of CNT-supported PdAgCu nanoparticle electrocatalysts: leaching lift-up and annealing fall-off. Electrochim Acta 293:149–159
Mazumder V, Chi M, Mankin MN, Liu Y, Metin Ö, Sun D, More KL, Sun Sh (2012) A facile synthesis of MPd (M = Co, Cu) nanoparticles and their catalysis for formic acid oxidation. Nano Lett 12:1102
Rand D, Woods R (1972) A study of the dissolution of platinum, palladium, rhodium and gold electrodes in 1M sulphuric acid by cyclic voltammetry. J Electroanal Chem 35:209–218
Bolzan AE, Arvia AJ (1992) The electrochemical behaviour of hydrous palladium oxide layers formed at high positive potentials in different electrolyte solutions. J Electroanal Chem 322:247–265
Shrestha BR, Nishikata A, Tsuru T (2012) Channel flow double electrode study on palladium dissolution during potential cycling in sulfuric acid solution. Electrochim Acta 70:42–49
Cadle SH (1974) Ring-disk electrode study of palladium dissolution. J Electrochem Soc 121:645–648
Hernandez F, Baltruschat H (2006) Electrochemical characterization of gold stepped surfaces modified with Pd. Langmuir 22:4877–4884
Pizzutilo E, Geigera S, Freakley SJ, Mingers A, Cherevko S, Hutchings GJ, Mayrhofer KJJ (2017) Palladium electrodissolution from model surfaces and nanoparticles. Electrochim Acta 229:467–477
Juodkazis K, Juodkazyte J, Šebeka B, Stalnionis G, Lukinskas A (2003) Anodic dissolution of palladium in sulfuric acid: an electrochemical quartz crystal microbalance study. Russ J Electrochem 39:954–959
Grdeń M, Łukaszewski M, Jerkiewicz G, Czerwiński A (2008) Electrochemical behaviour of palladium electrode: oxidation, electrodissolution and ionic adsorption. Electrochim Acta 53:7583–7598
Soszko M, Łukaszewski M, Czerwiński A (2020) Electrochemical dissolution of Pt-Pd-Ru alloys. J Electroanal Chem 877:Article 114547
Alvarez B, Berna A, Rodes A, Feliu JM (2004) Electrochemical properties of palladium layers on Pt(100). Surf Sci 573:32–46
Maksimov YM, Smolin AV, Podlovchenko BI (2007) On the ratio of processes of adsorbed oxygen layer formation and palladium surface layer dissolution at linear anodic potential sweep. Russ J Electrochem 43:1493–1498
Grdeń M, Kotowski J, Czerwiński A (1999) Study of electrochemical palladium behavior by the quartz crystal microbalance. I. Acidic solutions J Solid State Electrochem 3:348–351
Savadogo O, Lee K, Oishi K, Mitsushima S, Kamiya N, Ota K-I (2004) New palladium alloys catalyst for the oxygen reduction reaction in an acid medium. Electrochem Commun 6:105–109
Tarasevich MR, Bogdanovskaya VA, Kuznetsova LN, Modestov AD, Efremov BN, Chalykh AE, Chirkov YuG, Kapustina NA, Ehrenburg MR (2007) Development of platinum-free catalyst and catalyst with low platinum content for cathodic oxygen reduction in acidic electrolytes. J App Electrochem 37:1503–1513
Hubkowska K, Kubisztal J, Pająk M, Łosiewicz B, Czerwiński A (2021) Effect of the alloying metal on the corrosion resistance of Pd-rich binary alloys with Pt, Rh, and Ru in sulfuric acid. Materials 14:2923
Podlovchenko BI, Gladysheva TD, Maksimov YM, Volkov DS, Maslakov KI (2020) Specific features of the formation, catalytic activity, and corrosion stability of PdCu electrolytic co-deposit. J Solid State Electrochem 24:1439–1444
Tarasevich MR, Zhutaeva GV, Bogdanovskaya VA, Radina MV, Ehrenburg MR, Chalykh AE (2007) Oxygen kinetics and mechanism at electrocatalysts on the base of palladium–iron system. Electrochim Acta 52:5108–5118
Shao M-H, Kotaro S, Adzic RR (2006) Pd−Fe nanoparticles as electrocatalysts for oxygen reduction. J Am Chem Soc 128:3526–3527
Rusanova MYu, Tsirlina GA, Petrii OA, Safonova TYa, Vasil’ev SYu (2000) Palladium electrodeposits: dependence of structure and sorption properties on the deposition potential. Russ J Electrochem 36:457–464
Podlovchenko BI, Maksimov YuM, Gladysheva TD, Volkov DS (2021) Role of oxides in the electrochemical dissolution of Pd and its alloys. Mendeleev Commun 31:561–563
Kuznetsov VV, Telezhkina AV, Podlovchenko BI (2020) Electrolytic PdMo deposits with high corrosion resistance in relation to palladium. Mendeleev Commun 30:772–774
Trasatti S, Petrii OA (1992) Real surface area measurements in electrochemistry. J Electroanal Chem 327:353–376
Łukaszewski M, Soszko M, Czerwiński A (2016) Electrochemical methods of real surface area determination of noble metal electrodes – an overview. Int J Electrochem Sci 11:4442–4469
Swanson HE, Tatge E (1953) Standard X-ray diffraction powder patterns. US Department of Commerce. National Bureau of Standards Circular 539, Washington D.C
Baba K, Miyagawa U, Watanabe K, Sacamoto Y, Flanagan TB (1990) Electrical resistivity changes due to interstitial hydrogen in palladium-rich substitutional alloys. J Mater Sci 25:3910–3916
Rao CN, Rao KK (1964) Effect of temperature on the lattice parameters of some silver–palladium alloys. Can J Phys 42(7):1336–1342
Barbieri PF, de Siervo A, Carazzolle MF, Landers R, Kleiman GG (2004) XPS and XAES study of Ag–Pd and Cu–Ni alloys: spectra, shifts and electronic structure information. J Electron Spectroscopy and Related Phenomena 135:113–118
Selected Values of the Thermodynamic Properties of the Elements (1973) Hultgren R, Desai PD, Hawkins DT and others, Eds, Metals Park, Ohio, American. Soc Metals 778
Luef Ch, Al P, Flandorfer H, Kodentsov A, Ipser H (2005) Enthalpies of mixing of metallic systems relevant for lead-free soldering: Ag–Pd and Ag–Pd–Sn. J Alloys Compd 391:67–76
Podlovchenko BI, Kolyadko EA (1987) Effect of the nature of platinum group metals on the formation and properties of adatom silver coatings. J Electroanal Chem 224:225–235
Acknowledgements
Access to the Scanning Electron Microscopy facilities was granted by the Advanced Imaging Core Facility of Skoltech.
Funding
This work was supported by the Russian Foundation for Basic Research (project no. 19–03-00309) and in part by the M. V. Lomonosov Moscow State University Program of Development (the state assignment no. AAAA-A21-121011590088–4), financial support from Russian Science Foundation (No. 22–73-10198).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: The correct affiliation of Stanislav A. Evlashin is Skolkovo Institute of Science and Technology, Moscow, Russia.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Podlovchenko, B.I., Maksimov, Y.M., Gladysheva, T.D. et al. The effect of small silver inclusions on the palladium activity in formic acid oxidation reaction and corrosion stability. J Solid State Electrochem 27, 1767–1778 (2023). https://doi.org/10.1007/s10008-023-05404-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-023-05404-1