
Abstract
In this study, the standard molar Gibbs energy of formation of Y2Ru2O7(s) and Y3RuO7(s) was determined using calcia-stabilized zirconia (CSZ) as an electrolyte and air as a reference electrode. The cells can be represented by: (−)Pt/{Y2O3(s) + Y2Ru2O7(s) + Ru(s)}//CSZ//O2(p(O2) = 21.21 kPa)/Pt(+), (−)Pt/{Y3RuO7(s) + Y2Ru2O7(s) + Y2O3(s)}//CSZ//O2(p(O2) = 21.21kPa)/Pt(+). The electromotive force was measured in the temperature range from 981 to 1155 K and 932 to 1186 K, respectively. The standard molar Gibbs energy of formation of Y2Ru2O7(s) and Y3RuO7(s) from elements in their standard state was calculated by the least squares regression analysis of the data obtained in the present study and can be given, respectively, by: {ΔfG(Y2Ru2O7, s)/(kJmol−1) ± 2.22} = − 2554.1 + 0.625 ⋅ (T/K) and {ΔfG(Y3RuO7, s)/(kJmol−1) ± 2.45} = − 3249.5 + 0.635 ⋅ (T/K). The standard molar heat capacity Cop,m(T) of Y2Ru2O7(s) was measured using a heat flux–type differential scanning calorimeter (DSC) in the temperature range, from 307 to 780 K. The heat capacity was fitted into a mathematical expression and can be represented by: Cp, m(Y2Ru2O7, s, T)(JK−1mol−1) = 256.1 + 5.88 ∙ 10−2T(K) − 34.75 ∙ 105/T2(K). (307 ≤ T (K) ≤ 780). The heat capacity of Y2Ru2O7(s) was used along with the data obtained from the electrochemical cell to determine its decomposition temperature and stability in air and to calculate other thermodynamic parameters.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ruthenium-based oxides belong to the 4d series of transition metal oxides that show many interesting magnetic and electronic properties due to the enhanced radial extent of the 4d orbital [1, 2]. Electrons in ruthenates have an equal chance of displaying either localized behavior or iterant behavior dependent on structural or chemical influences [3]. Ruthenium shows unusual oxidation state ranging from + 2 to + 8 [4]. Mixed oxides containing both lanthanides and 4d transition elements have been the subject of an investigation by several researchers due to their unusual electronic and magnetic properties [5, 6]. Most of these studies have been largely limited to pyrochlore systems such as Ln2Ru2O7(s), where Ln is a rare earth element [7]. Rare earth ruthenium oxides wherein ruthenium is in the + 5 state were first synthesized by van Berkel and Ijdo [8]. Rossell described the orthorhombic structure as a superstructure of the cubic fluorite structure [9]. Rare earth ruthenates with composition Ln3RuO7 have been investigated extensively by Harada and Hinatsu with respect to their magnetic properties and low-temperature specific heat [10, 11]. Magnetic properties of La3RuO7(s) and La3OsO7(s) were also determined by Lam et al. [12].
In the Y-Ru-O system, the ternary compounds Y2Ru2O7(s) and Y3RuO7(s) are known. Cruickshank and Glasser [13] synthesized several rare earth platinum group double oxides and reported for the first time a Rooksby phase with the general formula RE3PGO7 ((PG) platinum group), including Y3RuO7(s). Though yttrium is not strictly a rare earth but has many characteristics in common with this group. In pyrochlores, when both A and B are transition metal cations the compounds are semiconductors. Thus, Ln2Ru2O7 (Ln = Pr/Lu) and Y2Ru2O7(s) are semiconducting with low activation energy [14]. Blacklock et al. measured the specific heats of the pyrochlore Y2Ru2O7(s) from 20 to 2 K. A Debye temperature of 499 K was calculated by them [15]. Taira and co-workers reported the magnetic properties of Y2Ru2O7(s) [16] and detected a magnetic transition at 80 K. Temperature-dependent optical properties were carried out by Lee et al. [17]. Pyrochlores as electrocatalysts for alkaline water electrolysis have recently been explored by Parrondo et al. [18]. Nanocrystalline yttrium ruthenate pyrochlores as new electrocatalysts for Zn-Air batteries was explored by Park et al. [19]. In a recent paper, Kim et al. studied the electrocatalytic properties of yttrium ruthenates and its stability in acidic medium for oxygen evolution reaction [20]. Shin and co-workers confirmed the formation of Y2Ru2O7(s) phase after trapping gaseous ruthenium at high temperature with yttria filter. In order to obtain information on operating conditions to efficiently trap ruthenium oxides that had volatilized from the DUPIC (Direct Use of spent PWR fuel in CANDU reactor) fabrication process, these experiments were carried out [21]. The determination of stability of the pyrochlore and its decomposition temperature is vital for developing process streams at the back end of the nuclear fuel cycle to recover precious ruthenium from radioactive waste. Understanding thermodynamic properties of these ruthenium oxides are of prime importance to determine their behavior in accident and high-temperature waste immobilization processes [22]. Thermodynamic data on the ternary oxides in the Y-Ru-O system is however lacking in literature.
In the present study, both the ternary oxides Y3RuO7(s) and Y2Ru2O7(s) in the Y-Ru-O system were synthesized and based on the phase relations, solid oxide-electrolyte galvanic cell measurements were carried out to determine the standard molar Gibbs energy of formation of Y2Ru2O7(s) and of Y3RuO7(s) in the temperature range from 981 to 1155 K and 932 to 1186 K, respectively. Heat capacity measurements of the pyrochlore Y2Ru2O7(s) was measured in the temperature range from 307 to 780 K using a differential scanning calorimeter (DSC-131) and its thermodynamic functions were evaluated from these experimental data.
Experimental
Materials
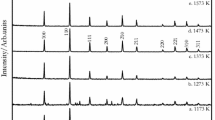
Y3RuO7(s) was synthesized from stoichiometric proportions of preheated Y2O3(s) (0.9985 mass fraction, Leico Industries Inc.) and RuO2(s) (0.997 mass fraction, Prabhat Chemicals, India). The oxide was intimately ground and the mixture was then pressed into pellets. The Y3RuO7(s) pellet was then subjected to a pressure of 50 MPa and then fired in air at 1373 K and furnace cooled to room temperature. The pellet was reground and repelletized several times to ensure homogeneity and a pure phase was obtained. The formation of the compound was confirmed by X-ray diffractometry using Cu-Kα radiation and is in agreement with those reported by van Berkel et al. [8]. Y2Ru2O7(s) was synthesized from stoichiometric proportions of preheated Y2O3(s) and RuO2(s) [23]. The mixture was pulverized and pelletized. Y2Ru2O7(s) pellets were sealed in an evacuated quartz ampoule and heated to T = 1400 K for several hours and cooled to room temperature. The formation of the compound was confirmed by X-ray diffractometry using Cu-Kα radiation, as shown in Fig. 1.

XRD pattern of Y2Ru2O7(s)
Cell design for e.m.f. measurements
Phase mixtures of {Y2O3(s) + Y2Ru2O7(s) + Ru(s)} and {Y3RuO7(s) + Y2Ru2O7(s) + Y2O3(s)} in the appropriate molar ratios were pressed into pellets of 10 mm in diameter and 3 mm thickness using a tungsten carbide lined die at a pressure of 100 MPa. The pellets were sintered at T = 1000 K for several hours. The phases in the sintered pellets remained unchanged after heat treatment. These pellets were then used for electromotive force (e.m.f.) measurement.
The oxide cell assembly
A double compartment cell assembly with 0.15-mol fraction calcia-stabilized zirconia (CSZ) solid electrolyte tube with one end closed and flat was used to separate the gaseous environments of the two electrodes. A schematic diagram of the in-house fabricated experimental set-up used for electrochemical measurements has been depicted in an earlier study [24]. Argon gas, with a flowing at a rate of 1 dm3 h−1, was purified by passing it through towers containing the reduced form of BASF catalyst, molecular sieves, and magnesium perchlorate which served as the gaseous atmosphere for the cell. The pellets for the working electrode were made by compaction and pelletization of appropriate phase mixtures as described earlier. The working electrode compartment was initially evacuated and flushed with argon gas. This procedure was repeated several times while simultaneously, the cell was heated to around 573 K so as to drive all moisture and other atmospheric gasses present in the electrode compartment. The argon gas served as the cover gas over the working electrode and was bubbled out at a steady rate through an oil bubbler without disturbing the equilibrium at the working electrode. An alumina guide tube directs the argon cover gas to the working electrode. Air was used as a reference electrode. Synthetic dry air from an air generator at atmospheric pressure was used for this purpose. A Faraday cage was placed between the furnace and cell assembly. The cage was grounded to minimize induced e.m.f. on the cell leads. Alumina sheathed platinum leads were used to measure the e.m.f. The temperature of the cell was measured by a calibrated chromel-alumel thermocouple located in the vicinity of the pellet. The e.m.f. of the cell was measured using a high impedance Keithley 614 electrometer. Voltages were reproducible in subsequent heating cycles. E.m.f. was measured after initially equilibrating the galvanic cells at 1000 K for several hours. The following cell configurations were employed in the present study:
-
Cell (I): (−)Pt / {{Y2O3(s) + Y2Ru2O7(s) + Ru(s)}//CSZ//O2(p(O2) = 21.21 kPa) / Pt(+)
-
Cell (II): (−)Pt / {{Y3RuO7(s) + Y2Ru2O7(s) + Y2O3(s)}//CSZ//O2(p(O2) = 21.21 kPa) / Pt(+).
The cells above were written in such a manner that the right-hand electrode is positive. The reversibility of the solid-state electrochemical cell was checked by micro-coulometric titration in both directions. A small quantity of current was passed through the cell in either direction. Removal of the applied current returned the cell e.m.f. to its original value. The e.m.f. of the cells was also found to be independent of flow rate of the inert gas passing over the sample electrode. The range of permissible oxygen partial pressures for purely ionic conduction for CSZ ranges from about 10−20 Pa at 1000 K to10−13 Pa at 1273 K as given by Pratt [25]; the oxygen partial pressure of these yttrium ruthenium oxides lie in this range.
Measurement of Heat Capacity of Y2Ru2O7(s)
Heat capacity measurements were carried out using a heat flux–type differential scanning calorimeter (Model: DSC-131, Setaram Instrumentation, France). The temperature calibration of the calorimeter was carried out in the present study by the phase transition temperature of the National Institute of Standards and Technology (NIST) reference materials. Heat calibration of the calorimeter was carried out from the enthalpies of transition of the reference materials as described in an earlier publication [26]. For the determination of heat capacity, NIST synthetic sapphire (SRM 720) in the powder form was used as the reference material. Heat capacity of the oxide was determined in the temperature range: 307 ≤ T (K) ≤ 780. The classical three-step method was used to determine the heat capacity in the continuous heating mode, in argon atmosphere at a heating rate of 5 K min−1 and a gas flow rate of 2 dm3 h−1. All three sets of experiment were performed under identical experimental conditions viz. heating rate, carrier gas flow rate, delay time, and temperature range. Two empty, flat-bottomed cylindrical aluminum crucibles of identical masses were selected for the sample and reference cells. In the first run, both the sample and reference cells were loaded with empty aluminum crucibles. The heat flow versus temperature was measured at a heating rate of 5 K min−1. In the second run, a known weight of NIST synthetic sapphire (SRM-720) was loaded in the sample cell keeping the crucible in the reference side empty and once again the heat flow versus temperature was measured in the same temperature range and at the same heating rate. In the third run, a known weight of the sample viz. Y2Ru2O7(s) was loaded in the sample cell, the reference cell being empty and the heat flow as a function of temperature was measured. About 300–350 mg of the sample was used for the heat capacity measurements.

Plot of e.m.f. as a function of temperature for the cell: (−)Pt / {Y2Ru2O7(s) + Y2O3(s) + Ru(s)}//CSZ//O2(p(O2) = 21.21 kPa) / Pt(+)
Theory
Solid-state electrochemical measurements using oxide cell
E.m.f. of the solid-state oxide electrochemical cell is related to the partial pressure of oxygen at the two electrodes and is given by the relation:
E is the measured e.m.f. of the cell in volts, R = 8.3144 J K−1 mol−1 is the universal gas constant, n is the number of electrons participating in the electrode reaction, F = 96485.3 C mol−1 is the Faraday constant, T is the absolute temperature, t(O2−) is the effective transference number of O2−ion for the solid electrolyte, and p″(O2) and p′(O2) are the equilibrium oxygen partial pressures at the positive and negative electrodes, respectively. The transport number of oxygen ion in the present electrolyte cell arrangement is near unity (t(O2−) > 0.99) at the oxygen pressures and temperatures covered in this study. Hence, the e.m.f. of the cell is directly proportional to the logarithm of the ratio of partial pressures of oxygen at the electrodes:
Thus,
where ln p′(O2) is the oxygen chemical potential over the positive electrode and RT ln p′(O2) is the oxygen chemical potential over the negative electrode.
Heat capacity measurements
The heat capacity of the sample under investigation using DSC-131 can be calculated by a simple comparison of the heat flow rates in three runs as illustrated in the literature [27]. For a defined step of temperature, the thermal effect corresponding to the sample heating is integrated. Thermal equilibrium of the sample is reached after each step of temperature. If Ti represents the initial temperature, the temperature interval step is chosen between Tj and Tj + 1, we define Tj = Ti + ΔT and Tj + 1 = Ti + (j + i) ΔT. The expression used for the calculation of heat capacity of the sample is given as:
where HFblank, HFRef, and HFsample represent the heat flow during the first, second, and third runs, respectively. Cp(Tj)sample and Cp(Tj)Ref represent the heat capacities of the sample and reference materials in J K−1 g−1 and Msample and MRef represent the mass of sample and reference, respectively. The heat capacity values thus obtained were then converted to J K−1 mol−1. The accuracy of measurements was checked by measuring the specific heat of Fe2O3(s) (mass fraction 0.998) in the temperature range from 307 to 700 K and the values were found to be within ± 3% as compared with the literature values [28].
Results and discussion
Δf G o (T) of Y2Ru2O7(s)
The reversible e.m.f. values of cell (I) measured as a function of temperature are shown in Fig. 2. The half-cell reaction at the cathode and the anode for the cell can be given by:

Plot of e.m.f. as a function of temperature for the cell: (−)Pt / {Y2Ru2O7(s) + Y2O3(s) + Y3RuO7(s)}//CSZ//O2(p(O2) = 21.21 kPa) / Pt(+)
The overall cell reaction can be represented by:
The least square regression analysis of the e.m.f. gives:
The uncertainties quoted are the standard deviation in e.m.f. The ΔrG°(T) for the reaction given in Eq. (7) involves the transfer of eight electrons and hence, from the Nernst equation, we get:
From Eqs. (7)–(9) and literature value of ΔfG°{Y2O3(s)} [29], ΔfG° of Y2Ru2O7 ( s) has been obtained as:
The error includes the standard deviation in e.m.f. and the uncertainty in the data taken from the literature. The Gibbs energy of formation is a linear function of temperature within the investigated temperature range. The intercept and the slope of this linear equation correspond, respectively, to the average values of the standard molar enthalpy and entropy of formation in the temperature range covered by e.m.f. measurement.
Δf G o (T) of Y3RuO7(s)
The reversible e.m.f. values of cell (II) measured as a function of temperature are shown in Fig. 3. The half-cell reaction at the cathode and the anode for the cell can be given by:
The overall cell reaction can be represented by:
The least square regression analysis of the e.m.f. gives:
The uncertainties quoted are the standard deviation in e.m.f. The ΔrG°(T) for the reaction given in Eq. (13) involves the transfer of two electrons and hence, from the Nernst equation, we get:
The value of ΔfGo (Y2Ru2O7, s) has been taken from Eq. (10), ΔfGo (Y2O3, s) from literature [28] and e.m.f. values from Eq. (14) and ΔfG° of Y3RuO7 (s) have been obtained as:
The errors include the standard deviation in e.m.f. values and the uncertainty in the data taken from the literature and the error due to the Gibbs energy of formation of Y2Ru2O7(s). The Gibbs energy of formation, given by Eq. (16), has a temperature independent term that gives the standard molar enthalpy and a temperature-dependent term that gives standard molar entropy of formation at the average experimental temperature.
Standard molar heat capacity of Y2Ru2O7(s)
The standard molar heat capacities of Y2Ru2O7(s) were obtained in the temperature range from 307 ≤ T (K) ≤ 780 and are presented in Table 1. The values of heat capacities are best fitted into the following mathematical expression by the least squares method.
The heat capacity value of Y2Ru2O7(s) at 298.15 K from the above equation was calculated to be 234.5 J K−1 mol−1. However, the heat capacity value of Y2Ru2O7(s) at 298.15 K estimated from heat capacity data of Y2O3 and RuO2 [30] by the Neumann-Kopp rule [31] was found to be 215.5 J K−1 mol−1. The molar heat capacity Cp,m given by the empirical Neumann-Kopp rule (NKR) gives a reasonable estimate of Cp,m in the vicinity of ambient temperature as mentioned by Leitner et al. [32] for most mixed oxides. However, Cp,m at constant pressure is a sum of several contributions:
where Cph is the contribution of lattice vibrations, Cdil for lattice dilation, and Cothers comprises contributions from conduction electrons, second-order phase transitions, formation of vacancies etc. [33]. Leitner et al. have further clarified that for complex oxides, which have magnetic ordering or electronic excitation, there will be deviation from the Neumann rule as in the present case. Kmiec et al. [34] have carried out magnetic properties of the Mott insulator Y2Ru2O7(s) and have observed that a non-linear local ordering of ruthenium magnetic moments occurs. This was also corroborated by Taira et al. [16], who measured the magnetic properties of the pyrochlore Y2Ru2O7(s) and noticed a magnetic transition at 80 K. Blacklock et al. [15] measured the specific heat below 20 and found a spin glass to antiferromagnetic transition. Taira et al. [35] observed a lambda-type specific heat anomaly at 77 K in the specific heat versus temperature plot and observed a significant jump in the temperature dependence of specific heat. Hence the discrepancy noted in the molar heat capacity values of Y2Ru2O7(s) determined by the present study versus those estimated by NKR.
Enthalpy of formation
The molar enthalpy of formation of Y2Ru2O7(s) at 298.15 K has been calculated by the second law method. Heat capacity data obtained in this study by using a differential scanning calorimeter along with heat capacity and transition enthalpies of Ru(s) [30], Y(s) [31], and O2(g) [36], and molar Gibbs energy of formation of Y2Ru2O7(s) obtained in this study using electrochemical cell, were used to determine the value of ΔfHo(Y2Ru2O7,s,298.15 K) which was found to be − 2602.4 kJ mol−1. The standard molar entropy S°(298.15 K) has been estimated by the author, by the Latimer method, which is from the entropy contribution of individual ions [31] which gives S°(298.15 K) = 245.2 (J K−1 mol−1). Based on the calculated value of entropy and the measured heat capacity, the thermodynamic functions of Y2Ru2O7(s) were calculated and the resulting values were extrapolated to 1000 K and given in Table 2 where fef denotes free energy function. The decomposition temperatures of Y2Ru2O7(s) and Y3RuO7(s) were calculated from the Gibbs energy data obtained in this study from Eqs. (10) and (16) and are 1778 K and 1760 K, respectively, in air. The decomposition temperature of RuO2(s) was calculated from the Gibbs free energy data of Cordfunke [37] and is 1647 K. The decomposition temperatures of Y2Ru2O7(s) and Y3RuO7(s) are higher than that of RuO2(s) at all partial pressures of oxygen and hence, these oxides will be stabler than ruthenium oxide.
Conclusion
Stability values of the ternary oxides, Y2Ru2O7(s) and Y3RuO7(s), were determined from Gibbs energy of formation data from elements in their standard state by using solid oxide electrochemical cells using calcia-stabilized zirconia as the solid electrolyte. The heat capacity data was fitted into a mathematical expression and can be represented by: Cop,m (Y2Ru2O7,s,T)(J K−1 mol−1) = 256.1 + 5.88 ∙ 10−2 T(K) − 34.75 ∙ 105/T2(K) in the temperature range from 307 to 780 K. The heat capacity of Y2Ru2O7(s) was reported for the first time and no other experimental data is available in the literature for comparison. The enthalpy of formation at 298.15 K of Y2Ru2O7(s) was determined. Phase diagram of higher order systems containing Y and Ru can be computed from these thermodynamic functions.
References
Wiss F, Raju NP, Wills AS, Greedan JE (2000) Structure and magnetism in Pr3RuO7. Int J Inorg Mater 2:53–59
Greedan JE (2001) Geometrically frustrated magnetic materials. J Mater Chem 11:37–53
Cao G, McCall S, Crow JE, Guertin RP (1997) Magnetic ordering and enhanced electronic heat capacity in insulating. Phys Rev Lett 78:1751–1754
Rard JA (1985) Chemistry and thermodynamics of ruthenium and some of its inorganic compounds and aqueous species. Chem Rev 85:1–39
Cava RJ (2004) Schizophrenic electrons in ruthenium-based oxides. Dalton Trans 10:2979–2987
Greedan JE, Sato M, Ali N, Datars WR (1987) Electrical resistivity of pyrochlore compounds R2Mo2O7 (R = Nd, Sm, Gd, Tb, Y). J Solid State Chem 68:300–306
Nishimine H, Wakeshima M, Hinatsu Y (2005) Structures, magnetic and thermal properties of Ln3MoO7 (Ln=La, Pr, Nd,Sm,Eu). J Solid State Chem 178:1221–1229
van Berkel FPF, Ijdo DJW (1986) The orthorhombic fluorite related compounds Ln3RuO7 Ln=Nd, Sm and Eu. Mater Res Bull 21:1103–1106
Rossell HJ (1979) Fluorite-related phases Ln3MO7, Ln = rare earth, Y or Sc, M = Nb, Sb, or Ta. J Solid State Chem 27:115–122
Harada D, Hinatsu Y (2002) Magnetic and calorimetric studies on one dimensional Ln3RuO7(Ln=Pr,Gd). J Solid State Chem 164:163–168
Harada D, Hinatsu Y (2001) A study of the magnetic and thermal properties of Ln3RuO7 (Ln=Sm,Eu). J Solid State Chem 158:245–253
Lam R, Wiss F, Greedan JE (2002) Magnetic properties of the fluorite related La3MoO7 phases M=Ru and Os. J Solid State Chem 167:182–187
Cruickshank KM, Glasser FP (1994) Rare earth platinum group mixed metal oxide systems. J Alloys Comp 210:177–184
Kobayashi H, Kanno R, Kawamoto Y, Kamiyama T, Izumi F, Sleight AW (1995) Synthesis, crystal structure, and electrical properties of the pyrochlores Pb2-x LnxRu2O7-y(Ln = Nd, Gd). J Solid State Chem 114:15–23
Blacklock K, White HW, Gurmen E (1980) Specific heats of the pyrochlore compounds Y2Mo2O7 and Y2Ru2O7. J Chem Phys 73:19661969
Taira N, Wakeshima M, Hinatsu Y (1999) Magnetic properties of ruthenium pyrochlores Y2Ru2O7 and Lu2Ru2O7. J Solid State Chem 144:216–219
Lee YS, Lee JS, Kim KW, Noh TW, Yu J, Takeda Y, Kanno R (2001) Optical investigation of A(2)Ru(2)O(7) (A = Y, Tl, and Bi): temperature dependent selfdoping effects. Physica C 364:632–635
Parrondo J, Morgan G, Capuano C, Ayersb KE, Raman V (2015) Pyrochlore electrocatalysts for efficient alkaline water electrolysis. J Mater Chem A 3:10819–10827
Park J, Park M, Nam G, Kim MG, Cho J (2017) Unveiling the Catalytic Origin of Nanocrystalline Yttrium Ruthenate Pyrochlore as a Bifunctional Electrocatalyst for Zn-Air Batteries. Nano Lett 17:3974–3981
Kim J, Shih PC, Tsao KC, Pan YT, Yin X, Sun CJ, Yang H (2017) High-performance pyrochlore-type yttrium ruthenate electrocatalyst for oxygen evolution reaction in acidic media. J Am Chem Soc 139:12076–12083
Shin JM, Park JJ, Shin SW, Kim KY (2005) Effectiveness of yttria filters for the removal of volatile ruthenium at high temperatures. Key Eng Mater 277:470–474
Kleykamp H (1988) The chemical state of fission products in oxide fuels at different stages of the nuclear fuel cycle. Nucl Tech 80:412–422
Kanno R, Takeda Y, Yamamoto T, Kawamoto Y, Yamamoto O (1993) Crystal structure and electrical properties of the pyrochlore ruthenate Bi2-xYxRu2O7. J Solid State Chem 102:106–114
Banerjee A, Singh Z, Venugopal V (2009) Heat capacity and Gibbs energy of formation of the ternary oxide CdRh2O4(s). J Solid State Ionics 180:1337–1341
Pratt JN (1990) Applications of solid electrolytes in thermodynamic studies of materials: a review. Metall Trans A 21:1223–1250
Banerjee A, Singh Z (2009) System Zn–Rh–O: Heat capacity and Gibbs free energy of formation using differential scanning calorimeter and electrochemical cell. J Solid State Electrochem 13:1201–1207
Sabbah R, Xu-wu AX, Chickos JS, Planas Leitao ML, Roux MV, Torres LA (1999) Reference materials for calorimetry and differential thermal analysis. Thermochim Acta 331:93–204
Hohne GWH, Hemminger WF, Flammershein HJ (2003) Differential scanning calorimetry, 2nd edn. Springer, Berlin
ASTD, Ver. 2.0, G. V. Belov, B. G. Trusov, 1983-1995, Moscow
Chase MW, Jr. JANAF Thermochemical Tables, Fourth edn., J Phys Chem, (monograph no. 91995)
Kubachewski O, Alcock CB, Spencer PJ (1993) Materials Thermochemistry, 6th. edn. Pergamon, Oxford
Leitner J, Vonka P, Sedmidubsky D, Svoboda P (2010) Application of Neumann-Kopp rule for the estimation of heat capacity of mixed oxides. Thermochim Acta 487:7–13
Grimvall G (1999) Thermophysical properties of materials, Elsevier, 367-368
Kmeic R, Swinkowska Z, Gurgul J, Rams M, Zarzycki A, Tomala K (2006) Investigation of the magnetic properties of Y2Ru2O7 by 99Ru Mössbauer spectroscopy. Phys Rev B 74:104425–104429
Taira N, Wakeshima M, Hinatsu Y (2000) Specific heat and ac susceptibility studies on ruthenium pyrochlores R2Ru2O7 (R = Rare Earths). J Solid State Chem 152:441–446
Graves K, Kirby KB, Rardin R (1991) FREED Version 2.1
Cordfunke EHP, Konings RJM (1988) The enthalpy of formation of ruthenium dioxide. Thermochim Acta 129:63–69
Acknowledgements
The authors wish to thank Dr. Meera Keskar, from FCD, BARC, for the XRD analysis. The authors are thankful to Dr. S. Kannan Head, Fuel Chemistry Division, and Dr. P. K. Pujari, Associate Director, R C, and I Group BARC, Mumbai, for their constant support and encouragement.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Banerjee, A. Determination of thermodynamic properties of the ternary oxides in the Y-Ru-O system by electromotive force measurements and differential scanning calorimetric measurements. J Solid State Electrochem 23, 1749–1755 (2019). https://doi.org/10.1007/s10008-019-04268-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-019-04268-8