
Abstract
A simple one-step electrodeposition without template allowed the synthesis of gold microstructures on a screen-printed carbon electrode. Chloroauric ions were reduced by applying a constant potential of − 0.6 V during 600 s. A preferential growth along the <111> directions produced leaf-like structures as confirmed by scanning electron microscopy and X-ray diffraction. The as-prepared Au microstructures worked as a support for tyrosinase immobilization allowing the preparation of a highly selective and sensitive biosensor for bisphenol A (BPA) detection. The cyclic voltammograms exhibit a well-defined anodic peak at 0.24 V in phosphate buffer solution (0.1 mol L−1, pH 7.0). The enzyme creates favorable conditions for the adsorption of BPA, and after 10 min of accumulation time, the calibration curve was linear in the range of 0.5–50 μmol L−1 with a detection limit of 77 nmol L−1 (S/N = 3) and a relative standard deviation (RSD%) of 0.54% (n = 10). Furthermore, the proposed biosensor displayed long-term stability and was successfully applied to determine BPA in spiked water samples.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Bisphenol A (BPA) has received considerable attention from the scientific community and regulatory agencies, becoming one of the most thoroughly investigated chemical substances in the last decades. More than 8 million pounds of this compound are produced each year for manufacturing epoxy resins and polycarbonate plastics as well as polyester, polysulfone, and polyacrylate resins [1,2,3]. Consequently, residues of this monomer can be present in numerous products, including food packaging materials, kitchenware, reusable water bottles, and in linings of canned food [4]. Thus, attention should be paid to the levels of BPA that migrates from these products to food and beverages, which represents the primary source of human exposure [5,6,7]. BPA is also present in others consumer products such as toys, electronics, dental sealants, sports safety equipment, eyeglass lenses, and medical devices (nasogastric tube, cardiopulmonary bypass, intravenous tubing, catheters) [8,9,10]. Therefore, continuous contact can also be through inhalation, dermal, or intravenous route [11]. Although the overall health issues attributed to BPA exposure are complex and controversial, the primary concern is related to its involvement in the pathogenesis of some endocrine disorders. Several studies indicated deleterious effects on the reproductive system, alterations in the brain development and memory function, metabolic disturbances including diabetogenic as well as obesogenic effects that increase the risk of cardiovascular diseases, alterations in the mammary gland development, and many others [12,13,14,15].
Additionally, human exposure to BPA is incredibly widespread considering the extensive usage of BPA-based polymers. To ensure safety levels of BPA in food and the environment, many analytical methods have been proposed, including high-performance liquid chromatography (HPLC) [16], gas chromatography-mass spectrometry (GC-MS) [17], high-performance liquid chromatography coupled to quadrupole time of flight mass spectrometry (HPLC-QTOF-MS) [18], and enzyme-linked immunosorbent assay (ELISA) [19]. These techniques are highly sensitive and accurate but often require expensive instrumentation, trained technicians, large sample volumes, and time-consuming extraction steps, restricting their applications.
On the other hand, electrochemical methods provide advantages of simple operation, cost-effectiveness, and fast response, becoming very attractive for BPA detection since this molecule poses two electroactive phenolic hydroxyl groups. However, modification of the electrode surface is mandatory to improve the performance of direct electrochemical analysis of BPA. For this purpose, nowadays, nanomaterials are widely used in the design of electrochemical sensors not only to improve sensitivity but also to lower the oxidation potential and prevent electrode surface fouling. For instance, various types of carbon-based nanomaterials including single and multi-walled nanotubes, graphite nanoparticles, and graphene exhibited excellent capabilities in the electrochemical oxidation with high sensitivity and selectivity [20,21,22]. The incorporation of gold nanostructures on the surface of the electrode also conferred high electrochemical activity and good performance for the detection of BPA [23,24,25]. Gold nanoparticles have excellent catalytic capacity and biocompatibility and are easy to prepare. Although most nanoparticles tend to grow in 0D structures, some conditions lead to the formation of 3D structures, such as dendrites and nanoflowers, which can significantly improve the sensitivity of the sensor.
Among many procedures available to synthesize three-dimensional architectures, electrodeposition is a useful and straightforward technique to grow gold nano/microstructures in an aqueous solution. Following this approach, in this work, gold was electrochemically deposited on a screen-printed carbon electrode to be applied as a platform to immobilize tyrosinase. This enzyme has already proved to be highly effective in the electrooxidation of BPA due to the presence of the copper ions in the active site of the protein [26,27,28,29]. In this study, however, the presence of the enzyme on the electrode increased the affinity for BPA adsorption, as confirmed by the dependence of the anodic peak intensity with the accumulation time. The proposed biosensor exhibited high sensitivity, reproducibility, and long-term stability.
Experimental part
Reagents and solutions
All the reagents used here were of analytical grade. Gold powder (99.99%, < 45 μm), potassium chloride (KCl, 99.0%), potassium ferrocyanide (K2[Fe(CN)6], 99.0%), 2,2-Bis(4-hydroxyphenyl) propane (Bisphenol A), Nafion® 117 solution (5% w/v), and tyrosinase enzyme extracted from mushrooms (lyophilized powder, ≥ 1000 unit/mg solid) were purchased from Sigma-Aldrich (Germany). Sodium dihydrogen phosphate anhydrous (NaH2PO4, 99.0%) and di-sodium hydrogen phosphate heptahydrate (Na2HPO4·7H2O, 98%), used in the preparation of the phosphate buffer solution, were obtained from Tedia Brazil. Aqueous solutions were prepared using ultrapure water collected from a Millipore Milli-Q purification system (resistivity > 18.2 MΩ cm at 25 °C). The preparation of the chloroauric acid (HAuCl4) solution consisted of dissolving the gold powder in aqua regia, and subsequent dilution to obtain a concentration of 6 g L−1 (30 mmol L−1). Finally, the lyophilized powder of the enzyme was dissolved in phosphate buffer solution 0.1 mol L−1, pH 7.0, at a concentration of 50 KU mL−1.
Materials and equipment
Screen-printed electrodes (SPCE), commercially available from DropSens (model DRP-C110), consisted of a carbon working electrode (4-mm diameter), a silver reference electrode, and carbon counter electrode. An Autolab (PGSTAT128N) potentiostat and NOVA 2.0.1 software was used for data acquisition in different electrochemical procedures. A pH meter of the brand MS Tecnopon Instrumentação (Mpa 210) was used for verification and adjusting the pH of the solutions. The scanning electron microscopy (SEM) images were recorded on Zeiss AURIGA FIB-SEM equipment, operating at a 15-kV acceleration voltage. The samples were fixed to the surface of an aluminum sample holder with double-sided carbon tape. X-ray diffraction analysis was accomplished by using a Rigaku D/max-2000 X-ray powder diffractometer, operated at 40 kV and 15 mA, using a monochromatic Cu Kα radiation (λ = 0.15184 nm). The diffraction data were collected among 2θ 8.0° and 80.0° with a 0.05° step size and counting time 1 s per step.
Electrode modification and biosensor preparation
First, gold was electrochemical deposited on the carbon electrode surface under potentiostatic conditions, by applying a constant potential of − 0.6 V during interval times of 150, 400, and 600 s. The electrolyte solution consisted of 0.1 mol L−1 Na2SO4 and 30 mmol L−1 HAuCl4. Afterward, the gold-modified screen-printed electrode (Au/SPCE) served as support for the immobilization of the tyrosinase enzyme (Tyr). Coating with Nafion® polymer was the procedure chosen to entrap the proteins on the surface. The biosensor, from now on Nafion/Tyr/Au/SPCE, was prepared by casting 10 μL of the enzyme solution (500 U) on Au/SPCE and allowed to dry at room temperature. Subsequently, 5 μL of the Nafion® solution was added, covering the entire surface to protect the enzyme, and avoid its leaching to the solution. After drying, the electrodes were preserved in 0.1 mol L−1 phosphate buffer (pH 7.0) at 4 °C. Au/SPCE and Nafion/Au/SPCE were also prepared for comparative purposes.
Results and discussion
Morphological analysis of electrodeposited gold
Figure 1a shows the SEM image of gold deposited on the screen-printed carbon electrode through electrolysis of chloroauric ions under a constant potential of − 0.6 V for 600 s. The experimental conditions favored the formation of leaf-like structures, of which most seems unattached, well-defined, and several micrometers long, but there are also small leaves growing on the top of the stem of the largest ones. By examining the micrograph at higher magnification, in the inset, revealed that the leaf morphology results from well-aligned and symmetric branches distributed on both sides of a long main trunk. The angles between the central stalk and the branches are around 68–70°, which is very close to the theoretical value of 67.81° found for preferential growth between two <111> directions in the plane (11\( \overline{2} \)) [30].

a Electron microscopy image of the leaf-like gold microstructures. b XRD diffraction of the Au/SPCE (black line) and SPCE (red line). Asterisk denotes non-identified peaks
To further understand the growth process, SEM images were recorded at 150, 400, and 600 s of reaction time. Figure 2 illustrates the time-dependent morphological evolution. Firstly, particles of irregular shapes grown horizontally cover most of the surface after 150 s of reaction time. There are also few stalks and small pointy sprouts spread, indicating anisotropic growth at this early stage. As the reaction proceeds, very short side branches emerge from the main trunk that further elongates until evolve to the leaf-like structures by increasing the reaction time to 600 s. It is worth mentioning that the gold electrodeposited on the SPCE has adopted less branched and straighter structures when compared to the hyperbranched Au dendrite produced on indium tin oxide (ITO) electrodes conducted under the same experimental conditions [31].

SEM images of the gold structures electrodeposited at different time: 150 s (a), 400 s (b), and 600 s (c) illustrating the morphology evolution
The Au/ SPCE X-ray diffraction pattern in Fig. 1b shows several peaks, mainly due to the SPCE substrate. Nevertheless, the peaks located at 38.2°, 44.5°, 64.6°, and 77.6° are attributed to [111], [200], [220], and [311] reflection planes of the face-centered cubic (fcc) structure of gold. The ratio of the intensities found for I(111)/I(200) is 4.0, which doubles the value commonly reported for polycrystalline Au (JCPDS 04-0784). This observation also suggests a preferential growth along the <111> direction.
Electrochemical analysis of Au/SPCE
The cyclic voltammogram of Au/SPCE in 0.5 mol L−1 sulfuric acid solution at the scan rate of 100 mV s−1 exhibited the gold oxidation peak and the corresponding stripping peak at 0.75 V and 0.44 V, respectively (data not shown). The charge passed in the gold oxide reduction peak allowed the calculation of the electrochemically active surface area, 0.15 cm2 was found [32]. For comparative purpose, the effective electrochemical area of bare SPCE was estimated to be 0.082 cm2 from the analysis of the ferri/ferrocyanide redox couple using the Randles-Sevcik equation [33].
Figure 3 shows the cyclic voltammograms of SPCE, Au/SPCE, Nafion/Au/SPCE, and Nafion/Tyr/Au/SPCE in 1 mmol L−1 potassium ferrocyanide with 0.1 mol L−1 KCl as the supporting electrolyte at a potential scan rate of 50 mV s−1. Comparing the voltammogram of the bare SPCE (green line) with the Au/SPCE (blue line), it is possible to infer that the leaf-like gold microstructures on the SPCE significantly improved the electrochemical response, giving a sharper and higher peak current, and shifting the peak potential about 125 mV towards less positive values. Furthermore, the peak-to-peak separation voltage (∆Ep) decreased from 166 to 68 mV which indicates an enhancement of the electron transfer rate. The absence of the redox couple at Nafion/Au/SPACE (red line) is due to the negatively charged sulfonic groups present in the Nafion that blocked the passage of the ferrocyanide molecules through the membrane. On the other hand, ferrocyanide has gained access in Nafion/Tyr/Au/SPCE (black line) due to some imperfections in the film, but the current intensities have diminished compared to Au/SPACE due to the adsorption of tyrosinase at the electrode surface. The baseline current also became higher in the presence of the enzyme.

Cyclic voltammograms at SPCE, Au/SPCE, Nafion/Au/SPCE, and Nafion/Tyr/Au/SPCE in 0.1 mol L−1 KCl containing 1 mmol L−1 K2Fe(CN)6 at 50 mV s−1
Electrooxidation of BPA
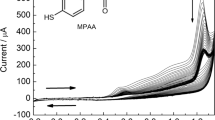
In order to verify the electrochemical performance of the biosensor for the determination of BPA, cyclic voltammograms were recorded in phosphate buffer solution at pH 7.0 at 20 mV s−1. As shown in Fig. 4a, no redox peaks appeared in the blank test; however, a sharp and irreversible peak arose by the addition of BPA in the electrolyte solution. In comparison to the Au/SPCE and Nafion/Au/SPCE, the current intensity was significantly higher at Nafion/Tyr/Au/SPCE. Furthermore, the anodic overpotential is reduced by ca. 30 and 80 mV, respectively. Unlike some other papers, where BPA acts as a substrate for tyrosinase, it is oxidized enzymatically, and subsequently, the reaction product is electrochemically reduced at the electrode surface [26,27,28,29]. Here, the presence of the enzyme created a favorable interface for the electrooxidation of BPA, improving the electrochemical activity. As reported by some studies, BPA can interact with some proteins and receptors mainly through hydrogen bonds and hydrophobic interactions [34, 35]. Figure 4b shows the schematic representation of the biosensor and the possible mechanism of the BPA electrooxidation.

a Cyclic voltammograms of Au/SPCE, Nafion/Au/SPCE, and Nafion/Tyr/Au/SPCE recorded in the absence (dotted line) and the presence of 30 μmol L−1 of BPA (solid line) in 0.1 mol L−1 phosphate buffer (pH 7.0). b Schematic representation of the Nafion/Tyr/Au/SPCE biosensor, and the possible mechanism of BPA oxidation at the electrode surface
According to the supplier’s recommendations (Sigma-Aldrich), the optimum pH for the enzyme tyrosinase from mushrooms is between 6.0 and 7.0. At pH 6.0, however, the oxidation potential shifted to more positive values (data not shown) which confirmed the involvement of a proton transfer in the electrode process. The most accepted mechanism for BPA electrooxidation consists in the transfer of two-electrons and two-protons [36]. Since a well-defined and reproducible signal was obtained at neutral pH, the further measurements were carried out at pH 7.0.
The next step was to determine the effect of the scan rate on the response of the biosensor. Figure 5a depicts cyclic voltammograms registered at scan rates ranging from 50 to 500 mV s−1. The anodic peak currents increased linearly, giving a regression equation of Ipa(mA) = 0.0071 + 0.6697 (V s−1) (R2 = 0.9975). This fact indicates that the oxidation of BPA is an adsorption-controlled electrode process. Furthermore, the peak potential shifted in the positive direction as the scan rate increased, this behavior is characteristic of an irreversible process.

a Cyclic voltammograms of 50 μmol L−1 BPA at scan rates from 50 to 500 mV s−1. Inset: dependence of the peak current on the scan rate. b Cyclic voltammograms of 20 μmol L−1 of BPA at preconcentration time from 0 to 11 min at 20 mV s−1. Inset: Dependence of the peak current on the accumulation time
For an adsorption-controlled process, the accumulation step is a simple and effective way to increase the sensitivity of the biosensor. Before the measurements, the biosensor was keep immersed in the 0.1 mol L−1 phosphate buffer solution, containing 10 and 20.0 μmol L−1 of BPA for 0–11 min to promote the adsorption of the molecule on the surface. Figure 5b shows the voltammetric responses in the presence of 20 μmol L−1 of BPA. The current increased almost proportionally as the accumulation time increased from 0 to 9 min when the surface reached saturation. The absence of the oxidation peak at time zero confirms the strong influence of the adsorption on the generation of the signal. The study conducted in 10 μmol L−1 displayed similar results. Thus, 10 min was the time chosen for subsequent measurements due to the higher sensitivity.
Analytical performance of Nafion/Tyr/Au/SPCE
The relationship between the peak currents and BPA concentrations was evaluated by registering cyclic voltammograms after successive additions of BPA from 0.5 to 90 μmol L−1. As shown in Fig. 6, the anodic peak gradually increased with the increase of the BPA concentration. The corresponding calibration curve shows a good linear response in the range of 0.5 and 50 μmol L−1 following the regression equation: Ip (μA) = 1.106 + 0.420 [BPA] (μmol L−1), R2 = 0.9930. The detection limit calculated according to S/N = 3 criterion is 77 nmol L−1.

Cyclic voltammograms of the biosensor in phosphate buffer solution (0.1 mol L−1, pH 7.0) containing different concentrations of BPA. Scan rate: 20 mV s−1. Inset: Dependence of the anodic peak current on the BPA concentration
Table 1 shows the performance of the Nafion/Tyr/Au/SPCE in comparison with other previously reported biosensors based on tyrosinase. Although the detection limit is higher or similar to those that employ different nanomaterials, this approach has the advantage of simple, fast, and easy preparation. Furthermore, the proposed biosensor reached good analytical performance in the determination of BPA by cyclic voltammetry analysis.
Selectivity, reproducibility, and stability
The response of the biosensor towards some potential interferents was measured to evaluate its selectivity for BPA. Glucose, ascorbic acid, and dopamine have no significant influence on the current response of BPA. Similarly, the addition of NaCl, KCl, and KNO3 did not interfere with the determination of BPA.
Regarding the reproducibility, the performance of three electrodes prepared following the same procedure gave a relative standard deviation of 11%. However, the RSD for ten successive measurements at the corresponding electrodes resulted as low as 0.5%, which suggests that the proposed biosensor has good repeatability.
The biosensor also showed excellent stability. It was found that after 6 months of storage the electrode still preserved 90% of its initial peak current response. This long-term stability suggests that no enzymatic reaction is involved in the oxidation process since it is no expected that the enzyme remains active for so long, as reported for most of the biosensors based on tyrosinase.
Recovery test
Finally, the practical application of the proposed biosensor was examined by a spike and recovery test in tap and mineral water samples. In this experiment, 0.5 μmol L−1 and 1 μmol L−1 of BPA were added to the solution consisting of 5 mL of the sample and 5 mL phosphate buffer (pH 7.0). As shown in Table 2, the mean recoveries of the spiked BPA samples were between 90.0 and 110%, and the relative standard deviations were in the range of 1.0 and 2.8%. These results indicate the potentiality of the developed biosensor for further detection of BPA in water samples.
Conclusion
The electrochemical reduction of chloroauric ions on the screen-printed carbon electrode resulted in the formation of well-defined leaf–like gold microstructures. These structures significantly improved the electron transfer reaction of potassium ferrocyanide and also served as a favorable platform for tyrosinase immobilization. In some way, the presence of the enzyme concomitantly with the gold morphologies created a beneficial environment for the adsorption of BPA. The resulted biosensor showed a wide linear range, low detection limit, excellent stability, and reproducibility as demonstrated by the cyclic voltammetry experiments performed at pH 7.0 in buffer phosphate solution. Moreover, the feasible preparation process and its long-term usability demonstrate the advantage of the proposed biosensor for BPA detection.
References
Reghunadhan Nair CP (2004) Advances in addition-cure phenolic resins. Prog Polym Sci 29(5):401–498
Lehmler HJ, Buyun L, Gadogbe M, Wei B (2018) Exposure to bisphenol A, bisphenol F, and bisphenol S in U.S. adults and children: the National Health and Nutrition Examination Survey 2013−2014. ACS Omega 3(6):6523–6532
Ramadan M, Sherman M, III RJ, Chaluvadi A, Swift L, Posnack NG (2018) Disruption of neonatal cardiomyocyte physiology following exposure to bisphenol-A. Sci Rep 8(1):7356–7367
U.S. Food & Drug Administration (2014) Bisphenol A (BPA): use in food contact application. https://www.fda.gov/newsevents/publichealthfocus/ucm064437.htm. Accessed 28 Mar 2019
Česen M, Lambropoulou D, Laimou-Geraniou M, Kosjek T, Blaznik U, Heath D, Heath E (2016) J Agric Food Chem 64(46):8866–8875
Torres A, Ramirez C, Romero J, Guerrero G, Valenzuela X, Guarda A, Galotto MJ (2015) Experimental and theoretical study of bisphenol A migration from polycarbonate into regulated EU food simulant. Eur Food Res Technol 240(2):335–343
Paseiro-Cerrato R, DeVries J, Begley TH (2017) Evaluation of short-term and long-term migration testing from can coatings into food simulants. J Agric Food Chem 5:2594–2602
Im J, Löffler FE (2016) Fate of bisphenol A in terrestrial and aquatic environments. Environ Sci Technol 50(16):8403–8416
Melcer H, Klecka G (2011) Treatment of wastewaters containing bisphenol A: state of the science review. Water Environ Res 83(7):650–666
Petrie B, Barden R, Kasprzyk-Hordern B (2015) A review on emerging contaminants in wastewaters and the environment: current knowledge, understudied areas and recommendations for future monitoring. Water Res 72:3–27
Huang R, Liu Z, Yuan S, Yin H, Dang Z, Wu P (2017) Worldwide human daily intakes of bisphenol A (BPA) estimated from global urinary concentration data (2000-2016) and its risk analysis. Environ Pollut 230:143–152
Chin KY, Pang KL, Mark-Lee WF (2018) A review on the effects of bisphenol A and its derivatives on skeletal health. Int J Med Sci 15(10):1043–1050
Vigui C, Mhaouty-Kodja S, Habert R, Chevrier C, Michel C, Pasquier E (2018) Evidence-based adverse outcome pathway approach for the identification of BPA as en endocrine disruptor in relation to its effect on the estrous cycle. Mol Cell Endocrinol 475:10–28
Beausoleil C, Emond C, Cravedi JP, Antignac JP, Applanat M, Appenzeller BR, Beaudouin R, Belzunces LP, Canivenc-Lavier MC, Chevalier N, Chevrier C, Elefant E, Eustache F, Habert R, Kolf-Clauw M, Magueresse-Battistoni BL, Mhaouty-Kodja S, Minier C, Multigner L, Schroeder H, Thonneau P, Viguie C, Pouzaud F, Ormsby JN, Rousselle C, Verines-Jouin L, Pasquier E, Michel C (2018) Regulatory identification of BPA as an endocrine disruptor: context and methodology. Mol Cell Endocrinol 475:4–9
Muñoz-de-Toro M, Markey CM, Wadia PR, Luque EH, Rubin BS, Sonnenschein C, Soto AM (2005) Perinatal exposure to bisphenol-A alters peripubertal mammary gland development in mice. Endocrinology 146(9):4138–4147
Háková M, Chocholou L, Havlíková S, Chvojka J, Solich P, Šatínský D (2018) An on-line coupling of nanofibrous extraction with column-switching high-performance liquid chromatography – a case study on the determination of bisphenol A in environmental water samples. Talanta 178:141–146
Cunha SC, Pena A, Fernandes JO (2015) Dispersive liquid–liquid microextraction followed by microwave-assisted silylation and gas chromatography-mass spectrometry analysis for simultaneous trace quantification of bisphenol A and 13 ultraviolet filters in wastewaters. J Chromatogr A 1414:10–21
Tian L, Lin L, Bayen S (2019) Optimization of the post-acquisition data processing for the non-targeted screening of trace leachable residues from reusable plastic bottles by high performance liquid chromatography coupled to hybrid quadrupole time of flight mass spectrometry. Talanta 193:70–76
Maiolini E, Ferri E, Pitasi AL, Montoya A, Di Giovanni M, Errani E, Girotti S (2014) Bisphenol A determination in baby bottles by chemiluminescence enzyme-linked immunosorbent assay, lateral flow immunoassay and liquid chromatography tandem mass spectrometry. Analyst 139(1):318–324
Li J, Kuang D, Feng Y, Zhang F, Liu M (2011) Voltammetric determination of bisphenol A in food package by a glassy carbon electrode modified with carboxylated multi-walled carbon nanotubes. Microchim Acta 172(3-4):379–386
Kannan PK, Hu C, Morgan H, Moshkalev SA, Rout CS (2016) Electrochemical sensing of bisphenol using a multilayer graphene nanobelt modified photolithography patterned platinum electrode. Nanotechnology 27(37):375504
Dong X, Qi X, Liu N, Yang Y, Piao Y (2017) Direct electrochemical detection of bisphenol A using a highly conductive graphite nanoparticle film electrode. Sensors 17(4):836–846
Manikandan VS, BalRam Adhikari B, Chen A (2018) Nanomaterial based electrochemical sensors for the safety and quality control of food and beverages. Analyst 143(19):4537–4554
Zhao WR, Kang TF, Lu LP, Shen FX, Cheng SY (2017) A novel electrochemical sensor based on gold nanoparticles and molecularly imprinted polymer with binary functional monomers for sensitive detection of bisphenol A. J Electroanal Chem 786:102–111
Wang A, Wei Y, Wang C (2015) Study on the Electrocatalytic oxidation of bisphenol A on au nanoparticles/carbon nanotubes composite modified electrode. J Anal Chem 70(1):67–71
Mita DG, Attanasio A, Arduini F, Diano N, Grano V, Bencivenga U, Rossi S, Amine A, Moscone D (2007) Enzymatic determination of BPA by means of tyrosinase immobilized on different carbon carriers. Biosens Bioelectron 23(1):60–65
Alkasir RSJ, Ganesana M, Won YH, Stanciu L, Andreescu S (2010) Enzyme functionalized nanoparticles for electrochemical biosensors: a comparative study with applications for the detection of bisphenol A. Biosens Bioelectron 26(1):43–49
Yin H, Zhou Y, Xu J, Ai S, Cui L, Zhu L (2010) Amperometric biosensor based on tyrosinase immobilized onto multiwalled carbon nanotubes-cobalt phthalocyanine-silk fibroin film and its application to determine bisphenol A. Anal Chim Acta 659(1-2):144–150
Kochana J, Wapiennik K, Kozak J, Knihnicki P, Pollap A, Woźniakiewicz M, Nowak J, Kościelniak P (2015) Tyrosinase-based biosensor for determination of bisphenol A in a flow-batch system. Talanta 144:163–170
Lin TH, Lin CW, Liu HH, Sheu JT, Hung WH (2011) Potential-controlled electrodeposition of gold dendrites in the presence of cysteine. Chem Commun 47(7):2044–2046
Ye W, Yan J, Ye Q, Zhou F (2010) Template-free and direct electrochemical deposition of hierarchical dendritic gold microstructures: growth and their multiple applications. J Phys Chem C 114(37):15617–15624
Trasatti S, Petrii OA (1992) Real surface area measurements in electrochemistry. J Electroanal Chem 321:353–376
Wang J (2000) Analytical electrochemistry. Wiley-VCH, 2nd Ed., New York
Li L, Wang Q, Zhang Y, Niu Y, Yao X, Liu H (2015) The molecular mechanism of bisphenol A (BPA) as an endocrine disruptor by interacting with nuclear receptors: insights from molecular dynamics (MD) simulations. PLoS One 0(3):e0120330
Hashimoto S, Shiomoto K, Okada K, Imaoka S (2012) The binding site of bisphenol A to protein disulphide isomerase. J Biochem 151(1):35–45
Lawrywianiec M, Smajdor J, Paczosa-Bator B, Piech R (2017) High sensitive method for determination of the toxic bisphenol A in food/beverage packaging and thermal paper using glassy carbon electrode modified with carbon black nanoparticles. Food Anal Methods 10(12):3825–3835
Acknowledgments
The authors thanks to INCTBio, PPGQ and Propesq/UFRGS. Also, to the Center of Electron Microscopy-CME/UFRGS and CNANO/UFRGS for the facilities used for the research. Filomeno thanks to CNPQ for his Ph.D. scholarship and Manoelly O. Rocha to CAPES for her MD Scholarship.
Funding
The authors received financial support from INCTBio (CNPq/INCT 465389/2014-7), CNPQ (Process: 550441/2012-3), CNPQ (Process: 190365/2014-5), Calouste Gulbenkian Foundation. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Finance Code 001
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Inroga, F.A.D., Rocha, M.O., Lavayen, V. et al. Development of a tyrosinase-based biosensor for bisphenol A detection using gold leaf–like microstructures. J Solid State Electrochem 23, 1659–1666 (2019). https://doi.org/10.1007/s10008-019-04252-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-019-04252-2