
Abstract
The influence of selected types of ammonium ionic liquid (AIL) additives on corrosion and functional parameters of lead-acid battery positive electrode was examined. AILs with a bisulfate anion used in the experiments were classified as protic, aprotic, monomeric, and polymeric, based on the structure of their cation. Working electrodes consisted of a lead-calcium-tin alloy utilized in the industry for manufacturing current collectors of positive electrodes in lead-acid batteries (LABs). This alloy was used in the first part of the study for the evaluation of corrosion intensity and stability of electrolyte with AIL additives. In the second part, the grid made of the examined alloy was covered with positive active mass, the composition of which was modified by selected AIL. The selection of an appropriate substance was made based on parameters such as corrosion potential, corrosion current, polarization resistance, and hydrogen and oxygen evolution potentials. Techniques such as linear sweep voltammetry, corrosimetry, and electrochemical impedance spectroscopy were used. The conducted measurements revealed that polymeric AIL additive had an exceptionally positive influence on the inhibition of the corrosion process in LAB and electrochemical window of the electrolyte. The influence of this compound on the specific capacity and resistance of the active mass was also discussed.

ᅟ
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Lead-acid batteries are secondary cells characterized by both high nominal potential (2.1 V) for a device with aqueous electrolyte and power density (123 W kg−1) [1, 2]. Their relatively good reliability and simple recycling made them a power supply, which can still compete with newer chemical power sources [1,2,3]. Despite many advantages, lead-acid batteries also have some weak points. The main drawbacks of lead-acid batteries include low specific energy, reaching only 40 Wh kg−1, and corrosion of current collectors (grids) made of lead alloys [4,5,6,7]. Corrosion affects mostly positive grid and thus causes shedding of the active mass out of its surface. In consequence, decrease of cell capacity occurs. Moreover, formation of corrosion products on the surface of the grid adversely influences its conductivity [8]. Thus far, gravimetric analysis of the lead alloy weight loss was the most common method for lead-acid battery corrosion analysis [9]. However, excursion peak measurements were reported as a potentially simpler and quicker procedure [9, 10]. When a more complex insight regarding the electrochemical behavior of the lead alloys is needed, electrochemical impedance spectroscopy can be used. This method provides data concerning changes on the grid-acidic electrolyte interface, which can be demonstrated in the form of Nyquist or Bode representations [11,12,13,14]. In addition to techniques which allow to analyze and measure the corrosion rate of lead alloys, there are a few known methods to reduce it. One of them is the addition of a corrosion inhibitor. Substances such as H3PO4, H3BO3, and several surfactants were successfully applied in lead-acid battery (LAB) for this purpose [1, 15, 16]. Recently, it has been found that addition of ionic liquids also decreases the corrosion rate [17,18,19].
Ionic liquids (ILs) have attracted the attention of researchers due to their possible use for various applications. This group of compounds received such interest due to their non-volatility, high thermal stability, and exceptional electrochemical properties [18]. Properties and applications of ILs are closely connected to their chemical structure. They can be designed easily by changing the type of anion or cation. Recently, one of their most promising applications is the enhancement of electrochemical performance of power devices [20,21,22,23]. Due to their electrochemical stability, ILs are widely used as electrolytes, especially in lithium-ion batteries or supercapacitors [24,25,26]. Furthermore, the influence of ILs on corrosion intensity has been widely confirmed. Various researchers show that addition of low doses of ILs can inhibit the corrosion of metals or alloys [27, 28], widen the potential window of the electrolyte [29], or act as a binder for electrode-active materials [30].
Ammonium-based ionic liquids (AILs) are one of the most popular groups of ILs. In the presented study, four compounds from this group were used to modify an electrochemical system consisting of a lead-calcium-tin alloy in 37 wt% sulfuric acid solution. As mentioned previously, the problem of grid corrosion concerns mainly positive electrodes; thus, the tests were focused exclusively on materials used for these elements. The aims of the experiments were, firstly, to investigate the effect of the AIL cation on the corrosion intensity of a specific lead-calcium-tin alloy and, secondly, to select one IL as a prospective additive to positive active mass.
Experimental section
Materials and synthesis
Hexyltrimethylammonium bromide (> 98%); N,N-dimethylhexylamine (98%); diallyldimethylammmonium chloride (65% aq.), poly(diallyldimethylammonium chloride) (M w < 100,000, 35% aq.), and sulfuric acid (98%) were purchased from Merck. Potassium hydroxide (> 85%) and methanol (> 99.8%) were delivered by Avantor Performance Materials.
Synthesis of aprotic ILs (AQA, MQA, PQA)
The aqueous solutions of diallyldimethylammmonium chloride and poly(diallyldimethylammonium chloride) were first evaporated under vacuum to obtain dry chlorides. The corresponding quaternary ammonium halide (0.01 M) was dissolved in methanol. Stoichiometric amount of potassium hydroxide in methanol was added. The reagents were mixed for 15 min under ambient conditions and then cooled to 0 °C. The precipitated inorganic salt was filtered off, and then, 0.01 mol of sulfuric acid was added. The neutralization was conducted for 5 min. The solvent was evaporated under vacuum, and the obtained product was dried in 60 °C under reduced pressure.
Synthesis of protic IL (HQA)
N,N-Dimethylhexylamine (0.01 M) was dissolved in methanol, and then, stoichiometric amount of sulfuric acid was slowly added. When all of the acid was added, the reagents were mixed vigorously for 5 min. The solvent was then removed under reduced pressure, and the obtained product was dried in 60 °C under vacuum.
The obtained compounds were classified based on the structure of the cation in their molecule as the following types: aprotic (AQA), protic (HQA), monomeric (MQA), and polymeric (PQA) (Table 1).
The electrolyte used for corrosion measurements was obtained by diluting sulfuric acid (98 wt% Merck) with the appropriate amount of distilled water to a concentration of 37 wt% (4.8 M), which is relevant to the concentration of the electrolyte in the fully charged battery. In the case of modified systems, specific quaternary ammonium ionic liquid in the amount of 5 mg per 1 cm3 of 37% sulfuric acid was added to the solution. The quantity of the additive was selected according to the results of previous experiments, in which a smaller amount of ionic liquid did not provide such satisfactory properties as 5 mg cm−3, while a greater amount led to the foaming of the electrolyte. For measurements of corrosion intensity and electrolyte properties, the lead-calcium-tin alloy strap was used as the working electrode. The composition of the alloy was the same as the positive grid produced by gravity casting. The counter electrode, with an approx. five times greater area compared to the working electrode, was made of pure lead (99.98% Pb, Avantor).
Preparation of positive electrodes for the capacity test consisted of three main stages. In the first step, the current collectors were produced by gravity casting. The obtained grid is shown on Fig. 1a. The second step was focused on covering the grid with active mass—pasting. Lead dust for active mass was produced by pot process in a Barton oxide system (Linklater reactor). Then, it was mixed with distilled water, sulfuric acid, and fibers in a laboratory kneader with two extruding stirrers. A small amount of AIL was added to the modified mass. Composition of the active mass is presented in Table 2. The paste was immediately applied on the grid, and the prepared plate was placed in ACS Sunrise climatic chamber (Italy) for the last step of process—curing. The curing program included two stages: 24 h at humidity greater than 95% followed by 24 h at humidity below 10%. The temperature was constant and kept at 60 ± 2 °C. The manufactured positive electrode before assembly is shown in Fig. 1b. Negative plates as counter electrodes in a 2-V cell were manufactured during a large-scale process under standardized industrial conditions in Autopart (Mielec, Poland).

Positive electrodes made of lead-calcium-tin alloy used for the study. a Positive grid manufactured by conventional gravity casting. b Plate pasted with positive active mass and cured for 48 h before assembly
NMR and elemental analysis of ILs
The NMR spectra were recorded using a Mercury Gemini 300 spectrometer with TMS as the internal standard operating at 300 MHz for 1H NMR spectra and 75 MHz for 13C NMR spectra. The carbon, hydrogen, and nitrogen contents were estimated using the elemental analysis by means of VARIO MICRO CUBE (Elementar Analysensysteme GmbH).
Hexyltrimethylammonium bisulfate (AQA): 1H NMR (300 MHz, CDCl3): δ 0.88 (t, 3H), 1.28 (m, 6H), 1.70 (m, 2H), 3.20 (m, 2H), 3.30 (s, 9H), 8.70 (s, 1H); 13C NMR (75 Hz, CDCl3): δ 14.0, 22.6, 23.9, 25.0, 31.4, 54.3, 66.8. Elemental analysis calc. (%) for C9H23NO4S (241.35): C 44.79, H 9.61, N 5.80; found: C 45.14, H 9.26, N 5.51.
N,N-dimethylhexylammonium bisulfate (HQA): 1H NMR (300 MHz, CDCl3): 0.88 (t, 3H), 1.28 (m, 6H), 1.71 (m, 2H), 2.85 (s, 6H), 3.24 (t, 2H), 6.90 (s, 1H), 8.30 (s, 1H); 13C NMR (75 Hz, CDCl3): δ 14.1, 22.5, 22.7, 26.0, 31.1, 43.4, 57.8. Elemental analysis calcd (%) for C8H21NO4S (227.32): C 42.27, H 9.31, N 6.16; found: C 41.89, H 9.03, N 5.89.
Diallyldimethylammonium bisulfate (MQA): 1H NMR (300 MHz, D2O): 3.00 (s, 6H), 3.96 (m, 4H), 5.60 (m, 4H), 6.05 (m, 2H); 13C NMR (75 Hz, D2O): δ 49.0, 65.0, 122.4, 127.7. Elemental analysis calcd (%) for C8H17NO4S (223.29): C 43.03, H 7.67, N 6.27; found: C 43.29, H 8.00, N 6.55.
Poly(diallyldimethylammonium bisulfate) (PQA): 1H NMR (300 MHz, D2O): 1.30 (m, 4H), 2.69 (m, 2H), 3.12 (s, 6H), 3.78 (m, 4H); 13C NMR (75 Hz, D2O): δ 29.4, 41.1, 41.5, 73.2. Elemental analysis calcd (%) for (C8H17NO4S)n (223.29): C 43.03, H 7.67, N 6.27; found: C 43.39, H 7.65, N 5.99.
Thermal analysis of ILs
Thermal transition temperatures were determined by DSC, with a Mettler Toledo Stare DSC1 (Leicester, UK) unit. The DSC1 apparatus was calibrated by measuring the following standards: In (purity 99.999%, T m = 156.49 °C, ΔH = 29.38 J g−1), Pb (purity 99.99%, T m = 327 °C, ΔH = 22.25 J g−1), and Zn (purity 99.998%, T m = 418.78 °C, ΔH = 106.53 J g−1). ILs (between 5 and 6 mg) were placed in aluminum pans and heated from 25 to 120 °C at a heating rate of 10 °C min−1, cooled with an intracooler at a cooling rate of 10 °C min−1 to −100 °C, then heated again to 120 °C under nitrogen (99.998% purity) with flow rate of 50 mL min−1.
Thermogravimetric analysis was performed using a TGA/DSC1 Mettler Toledo apparatus. The thermal analysis apparatus was calibrated by measuring the following standards: In (purity 99.999%, T m = 156.49 °C, ΔH = 29.38 J g−1), Pb (purity 99.99%, T m = 327 °C, ΔH = 22.25 J g−1), Zn (purity 99.998%, T m = 418.78 °C, ΔH = 106.53 J g−1), and Al (purity 99.99%, T m = 660 °C, ΔH = 340.15 J g−1). The thermogravimetric measurements were performed at a heating rate of 10 °C min−1 from 25 to 550 °C under dynamic nitrogen atmosphere (99.998% purity, flow rate of 50 mL min−1), using sample of approx. 5–8 mg in an open aluminum pan.
Thermogravimetric analysis was performed three times on material samples retrieved from different production batches in order to confirm the reproducibility of the synthesis.
Elemental ICP analysis
Elemental ICP analysis of lead-calcium-tin alloy was performed by means of AGILENT 725 ES ICP optical emission spectrometer. X-ray diffraction of lead powder samples was performed to analyze the precursor of active mass in terms of its physicochemical parameters in order to be used in lead-acid battery technology. X-ray diffractometer Philips PW1050, with Cu Kα radiation and filter Ni, was used. Measurements were carried out in 2θ range of 5°–50° at a scan step of 0.04°.
Electrochemical measurements
The conductivity of the prepared electrolytes was measured by direct-current method with a laboratory conductometer Mettler Toledo SevenCompact S230 (Leicester, UK).
Electrochemical stability of the electrolyte was analyzed by linear sweep voltammetry technique (LSV) performed at a scan rate of 10 mV s−1.
The experiment on corrosion intensity of the alloy in modified electrolyte was a combination of electrochemical impedance spectroscopy (EIS), corrosimetry (CM), and LSV. EIS was carried out within a frequency range of 50 mHz to 10 kHz at open-circuit voltage (OCV). The number of points from the initial to the final frequency was 200, and the value of the sinus amplitude V rms was ± 7 mV. CM was performed at a scan rate of 0.167 mV s−1 within a potential range of ± 25 mV versus corrosion potential. The LSV measurements were conducted at a scan rate equal to 0.2 mV s−1, with potential range from − 180 to + 180 mV versus open-circuit voltage. The measurements were repeated to confirm whether the corrosion parameters change with time. OCV was kept for 5 h between these two experimental series to ensure that the cell was at a thermodynamically steady state before the subsequent measurements. The measurements were performed at 25 °C in three electrode configurations with a mercury/mercurous sulfate (Hg/Hg2SO4 in 1 M H2SO4) reference electrode. The geometric area of the circle-shaped working electrode was 1.0 cm2. Before each experiment, the lead alloy working electrode was mechanically cleaned with sandpapers of grit sizes P2500 and P5000, degreased with acetone, and etched with acetic acid. After every step of cleaning, the electrode surface was washed with distilled water. All corrosion measurements were performed using VMP3 (BioLogic®, France) multichannel potentiostat/galvanostat with EC-Lab® software.
The capacity test of the reference (BASIC) and modified positive plates was performed in 2-V cells. Each plate (working electrode) was assembled between two negative electrodes, soaked in electrolyte (sulfuric acid solution with a density of 1.24 g cm−3) and formed afterwards. The formation process and capacity testing were carried out using a five-step program (Table 3). The temperature value during the formation process did not exceed 60 °C. After formation process, cyclic stability of assembled cells during 20 charge/discharge cycles was examined with discharge current of 350 mA, stopped at 1.75 V, and charged with maximal current of 700 mA. Electrochemical impedance spectroscopy was carried out within a potential range from 10 mHz to 1 kHz at open-circuit voltage. The number of points per decade was equal to 6, and the value of the sinus amplitude V rms was ± 7 mV. Electrochemical measurements were performed using VMP3 (BioLogic®) multichannel potentiostat/galvanostat with 20-A current booster.
Results and discussion
The bisulfates were obtained with high yields, exceeding 90%. The structures of the compounds were confirmed by NMR spectra. The protons in the aliphatic substituent (AQA, HQA) generated signals at 0.88 ppm for methyl groups and 1.28 ppm for methylene groups. Methylene groups in position β to the charged nitrogen were noted at 3.20 ppm (AQA) and 3.24 ppm (HQA). Protons in position α to the quaternary nitrogen atom generated signals at 3.20 ppm (AQA) and 3.24 ppm (HQA). Aliphatic methylene groups in polymeric IL (PQA) were observed as signals at 1.30 ppm. Methine groups in PQA were observed at 2.69 ppm. Signals at 3.78 ppm (PQA) and 3.96 ppm (MQA) were identified as protons in methylene groups in position α to the nitrogen atom. Protons in double bonds of monomeric IL (MQA) were observed at 5.60 ppm for methylene and 6.05 ppm for methine groups. Signals of methyl groups bonded directly to the quaternary nitrogen occurred as strong signals between 2.85 and 3.30 ppm. The measured amount of carbon, hydrogen, and nitrogen atoms in the samples did not differ by more than ± 0.4% from the calculated values.
All synthesized compounds were classified as ionic liquids and investigated in terms of phase transitions and thermal stability (Table 4).
Hexyltrimethyl ammonium bisulfate (AQA) exhibited glass transition at – 40 °C, crystallization at − 7 °C, and melting at 44 °C. Substitution of one methyl group with a proton (HQA) resulted in a decrease of glass transition value to − 69 °C. In the case of the diallyldimethylammonium cation (MQA), only glass transition was observed at − 67 °C; however, no phase transitions were noted in the range from − 160 to + 160 °C for the polymeric form of this cation (PQA).
The studied bisulfates were stable up to approx. 200 °C (Fig. 2). The hexyltrimethylammonium cation started to decompose (T i) at 222 °C, and maximum decomposition (T max) occurred at 332 °C. The protic IL (HQA) was characterized by lower stability, with maximum decomposition at 293 °C. Both aprotic (AQA) and protic IL (HQA) exhibited multistep decomposition. The first step occurred in the range of 221 to 273 °C with 10% mass loss and the second from 273 to 363 °C with 84% mass loss for aprotic IL (AQA). In the case of the protic IL (HQA), the first step is dominant. Approx. 90% of mass loss occurred in the range of 193 to 376 °C, while the second occurred from 376 to 462 °C with 3% of decomposition. The diallyldimethylammonium bisulfate (MQA) decomposed at 185 °C with maximum decomposition at 302 °C, while its polymeric form (PQA) was much more thermally stable with initial and maximum decompositions at 245 and 295 °C, respectively. The first decomposition step of diallyldimethylammonium bisulfate (MQA) occurred in the range from 185 to 271 °C with 15% of mass loss, while the major step (44%) ranged between 271 and 332 °C. For the polymeric IL (PQA), the first decomposition step (40%) occurred from 224 to 342 °C and the second from 324 to 383 °C, with 12% of mass loss. The thermogravimetric curves of three PQA samples are presented in Fig. OR_1 (in Online Resource 1). Each sample is a representative of different synthesis batches. The curves overlap each other, and the highest difference between them reached 2 percentage points, which proves that the synthesis was carried out correctly with proper subsequent preparation of the product.

TGA measurements of AILs
Composition of the lead-calcium-tin alloy, which was used to check the electrochemical stability of electrolytes and corrosion intensity, is presented in Table 5. The concentration of each component matched the composition characteristic of a standardized alloy for LAB electrodes. Lead, tin, and calcium were the three main components. Other elements comprised approx. 0.02 wt% of the sample.
Measurements of electrochemical stability of the electrolyte, presented in Fig. 3, showed that addition of IL influenced oxygen and hydrogen evolution potentials. The measurements also indicated that there are no side reactions resulting from, for example, the decomposition of ionic liquids. Oxidation and reduction peaks at the potential value of − 1.0 V versus Hg/Hg2SO4 resulted from oxidation and reduction reactions of lead. Additionally, the values of electrolyte conductivity, presented in Table 6, indicated that the most prominent effect can be observed when PQA was used as a modifying compound. The conductivity of that solution was the highest among the modified materials and similar to the BASIC. Moreover, the polymeric additive contributed to an enhancement of electrolyte stability, which is desirable in lead-acid battery application.

Electrochemical window of unmodified environment and electrolytes with 5 mg cm−3 AIL additive; used technique was linear sweep voltammetry at 10 mV s−1
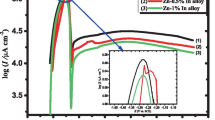
Corrosion parameters and polarization resistances are presented in Table 7. Extrapolation of corrosion potential and current density from the Tafel plot is presented in Fig. OR_2 (in Online Resources 1). The unmodified system (marked as BASIC) was stable during both series of measurements. Corrosion proceeded approximately at the constant rate. Measurements carried out for modified electrolytes showed that two types of additives, MQA and PQA, generated significant changes in the electrode behavior. In both cases, the corrosion potential increased from approx. − 1030 mV to approx. − 800 mV and corrosion current sharply decreased in the second series of measurements performed after 5 h of rest. Polarization resistance also increased rapidly, achieving over 10,000 times higher value than in the case of the BASIC electrolyte. This indicates that a layer was formed on the surface of the lead alloy. This layer was durable and adhesive enough to prevent the further corrosion of the alloy, and, most importantly, it did not affect the reactions occurring on the electrode. The structure of AIL molecules suggested the formation of an adsorbing layer. The type of cation in MQA and PQA indicated that thin organic film was formed on the surface of the alloy. Corrosion in the electrolyte with the HQA additive was characterized by stable corrosion potential and moderate changes in corrosion current and polarization resistance. Electrolyte containing AQA showed a different behavior. Corrosion potentials resulting from corrosimetry varied from − 1041 mV in the first series of measurements to − 1012 mV in the second series. Additionally, polarization resistance was over 1000 times higher than in the pure electrolyte. On the other hand, the corrosion potentials in linear sweep voltammetry were stable at − 1041 mV during measurements. Corrosion current density decreased sharply in comparison to the BASIC system while polarization resistance reached a 600 times higher value; thus, the reduction of corrosion current values from LSV was much lower. The most probable explanation for this phenomenon was the instability of the surface layer on the lead-calcium-tin alloy. The formed layer, similar to that in the case with MQA and PQA, reduced the corrosion of current collectors, but it was not stable enough to successfully protect the alloy in case of larger changes of potential. During LSV, the potential changes were too high; hence, this layer was partially destroyed. During CM, only slight fluctuations of potential near the corrosion potential value were applied. In this case, the layer formed on the electrode surface gained higher durability.
Tafel plots (Fig. 4) and Bode plots (Figs. 5 and 6) of the examined systems at the beginning of the test (Figs. 4a and 5, respectively) and after 5-h resting time (Figs. 4b and 6, respectively) enabled further analysis of corrosion process. The Bode plot of BASIC system shows that initially, an electrical double layer formed on the electrolyte/electrode interface. After 5 h of immersion in the electrolyte, a low conductive PbSO4 layer covered the electrode. At low frequencies, the process is controlled by diffusion. The slight increase of anodic potential and current in the Tafel plot indicated that the longer the process runs, the more energy is needed for the PbSO4 layer to overbuild on the surface of the electrode. AQA additive increased the initial resistance of the system. A possible explanation includes the occurrence of a reaction with the ionic liquid. Prolonged immersion of the electrode in the modified electrolyte resulted in the appearance of two time constants. The first corresponds to the charge transfer process, in which the resistance increased during measurements. The second time constant is related to the diffusion through the layer on the electrode surface, in which the resistance was greater than that of the BASIC system. Increase of the measured resistance can be attributed to substitution of the hydrophobic carbon chain into the surface layer. On the other hand, addition of HQA to the electrolyte seems to have a different effect. The layer formed on the surface at the beginning of the experiment was thinner than in the previous case. Thus, the diffusion process through this layer proceeded with greater intensity. Bode plots of the MQA modified system were characterized by one time constant. The measured resistance was undeniably higher than previously. This phenomenon can be associated with the structure of the MQA molecule. This compound might have been incorporated in the surface layer, which caused significant resistance during the charge transfer process. PQA can be a steric hindrance due to its size. Its influence on the charge transfer resistance was lower compared to MQA. This suggests that the formation of the layer inhibits the corrosion process rate but has smaller impact on the electrode conductivity.

Tafel plots of lead-calcium-tin alloy in electrolytes used in the study; scan rate 0.01 mV s−1, three-electrode cell. a Measurements directly after assembly of the cell and b after 5-h rest

Bode plots of lead-calcium-tin alloy in the examined electrolytes; frequency range from 50 mHz to 10 kHz, three-electrode cell, first series of measurements

Bode plots of lead-calcium-tin alloy in the examined electrolytes; frequency range from 50 mHz to 10 kHz, three-electrode cell. Measurements after 5 h of rest
The measurements carried out on a model electrochemical system were used as a background for selecting one AIL as a prospective additive to the lead acid battery paste. A small amount of PQA proved to affect the examined electrochemical system in a clearly positive way. Its good corrosion inhibition parameters, wide range of electrolyte stability, and high conductivity were superior to other additives used in this study. This justified the selection of PQA as an additive to positive active mass.
The active mass was obtained from lead powder made in a Barton pot. XRD analysis of lead dust showed that the used material consisted of 71.4% α-PbO, 4.6% β-PbO, and 24.0% Pb, in relative percent. This composition confirmed that the physicochemical parameters were appropriate for use in the lead-acid battery industry.
Charge curves of lead-acid cells (Fig. 7a) show that the charging process of cells with BASIC and modified positive plates proceeded in a similar manner. Only slightly prolonged charging time can be observed for cells with a modified positive plate. It can be observed that the potential of the electrode was slightly lower during discharge (Fig. 7b) in comparison to the cell containing a plate without PQA; however, this phenomenon did not have any negative influence on the capacity. In order to evaluate how the modification of positive mass influenced the performance of the lead-acid cell during a longer period of time, 20 consecutive galvanostatic cycles were performed (Fig. 8). As can be noted, the cell with positive electrode modified by ionic liquid showed better capacity retention than the reference sample. It can be observed that positive plates with IL additive have higher capacity than the reference group (approx. 8% higher in the first and third cycles). Capacity of positive BASIC plates and those modified by PQA addition increased between the first and second discharges by approx. 24 and 2%, respectively. A much lower increase of capacity during the first cycles indicated a better formation of the electroactive material. Ionic liquid enhanced the lead-acid cell performance not only at the beginning, but also throughout numerous cycles, which suggests that no negative reaction (such as ionic liquid oxidation) occurred on the positive electrode in the modified cell. A more detailed explanation including additional figures which depict the grid before and after galvanostatic cycling can be found in the Online Resources 1 (Fig. OR_3–OR_5). Analysis of impedance spectroscopy data (Fig. 9) showed that charge transfer resistance was visibly lower for the modified electrode. The average value for three plates varied from 1.51 μΩ (modified active mass) to 2.31 μΩ (BASIC one). Bode plots also indicated that no additional layer formed on the surface between the active mass and the grid.

a Charge and b discharge curves of lead-acid cells with C10 current

Capacity of examined cells during 20 galvanostatic cycles

Bode plots of BASIC and modified positive plates after formation; frequency range from 10 mHz to 1 kHz
Conclusions
The lead-acid battery electrolyte and active mass of the positive electrode were modified by addition of four ammonium-based ionic liquids. In the first part of the experiment, parameters such as corrosion potential and current, polarization resistance, electrolyte conductivity, and stability were studied. Data from the measurements allowed to select one AIL which ensured the most optimal alteration of the studied parameters. The second part was focused on performance tests of active mass modified by the addition of the previously selected AIL.
It could be observed that the added amount of AIL affected the performance of the electrochemical systems. The obtained results indicate that the type of the cation in the molecule of AIL has notable influence on the properties of these compounds and thus on the operational parameters of the modified electrochemical systems. PQA showed the most promising influence; therefore, this IL was selected as an effective additive for capacity tests of the positive electrode. Significant decrease of corrosion rate of the positive electrode in the modified system was observed. Aside from the decrease of the value of corrosion current, a shift in the corrosion potential by more than 200 mV was also observed. The formation of an adsorbing layer on the surface of the electrode is the most plausible explanation of this phenomenon. This layer may inhibit corrosion intensity without affecting the electron transfer process between the active material and current collector. The structure of the molecule may support the hypothesis associated with the formation of an organic layer. Furthermore, PQA improved the electrochemical stability of the electrolyte, which is an important factor during battery charging (as it leads to a decreased rate of electrolyte decomposition). After introduction of PQA to the active mass in the second part of the tests, an enhancement of electroactive material capacity could be observed, despite a small decrease of the initial potential of the electrode. An additional advantageous result is that its resistance was approx. two times lower than that of the unmodified material.
References
Pavlov D (2011) Lead-acid batteries: science and technology. Elsevier, Amsterdam
Moseley PT, Garche J, Parker CD, Rand DAJ (2004) Valve-regulated lead-acid batteries. Elsevier, Amsterdam
Taguchi M, Hirasawa T (2000) Influence of Sn content on the corrosion behavior of Pb-Ca-Sn alloys in high-temperature sulfuric acid solution. Mater Trans JIM 41:1307–1311
Dimitrov M, Pavlov D, Rogachev T, Matrakova M, Bogdanova L (2005) Processes taking place in the paste of lead-acid battery plates during soaking prior to formation and their influence on battery performance. J Power Sources 140:168–180
Jung J, Zhang L, Zhang J (2016) Lead-acid battery technologies: fundamentals, materials and applications. CRC Press, Boca Raton
Prengaman RD (1995) Wrought lead-calcium-tin alloys for tubular lead/acid battery grids. J Power Sources 53:207–214
Osório WR, Bortolozo AD, Peixoto LC, Garcia A (2014) Mechanical performance and microstructure array of as-cast lead-silver and lead-bismuth alloys. J Power Sources 271:124–133
Bui N, Mattesco P, Simon P, Steinmetz J, Rocca E (1997) The tin effect in lead-calcium alloys. J Power Sources 67:61–67
Lach J, Czerwiński A (2016) The anodic excursion peak as a rapid way of measuring the corrosion of lead alloys. Int J Electrochem Sci 11:9355–9368
Lach J, Obrębowski S, Czerwiński A (2015) Origin of the “Excursion Peak” during cycling voltammetry of Pb-Sn alloys. J Electroanal Chem 742:104–109
Osório WR, Peixoto LC, Garcia A (2015) Electrochemical and mechanical behavior of lead-silver and lead-bismuth casting alloys for lead-acid battery components. Metall Mater Trans A 46:4255–4267
El-Sayed AR, Ibrahim EMM, Mohran HS, Ismael M, Shilkamy HAS (2015) Effect of indium alloying with lead on the mechanical properties and corrosion resistance of lead-indium alloys in sulfuric acid solution. Metall Mater Trans A 46:1995–2006
El-Sayed AR, Mohran HS, Shilkamy HAS (2016) Effect of phosphoric acid concentration on conductivity of anodic passive film formed on surface of lead-indium alloy. Trans Nonferrous Met Soc China 26:882–894
Duan Y, Li P, Zhang M, Peng M, Ma L, Shu B (2017) Corrosion behavior of Pb-39Mg-10Al-1.5B alloy in sodium halide solutions. J Alloys Compd 729:1108–1117
Bullock KR (1979) The effect of phosphoric acid on the positive electrode in the lead-acid battery. J Electrochem Soc 126:360–365
Garche J, Döring H, Wiesener K (1991) Influence of phosphoric acid on both the electrochemistry and the operating behavior of the lead/acid system. J Power Sources 33:213–220
Rezaei B, Taki M (2008) Effects of tetrabutylammonium hydrogen sulfate as an electrolyte additive on the electrochemical behavior of lead acid battery. J Solid State Electrochem 12:1663–1671
Ohno H (2001) Electrochemical aspects of ionic liquids. Wiley, Tokio
Torriero AAJ (2015) Electrochemistry in ionic liquids vol. 2: applications. Springer, Cham
Frackowiak E, Lota G, Pernak J (2005) Room-temperature phosphonium ionic liquids for supercapacitor application. Appl Phys Lett 86:164104
Rezaei B, Ensafi AA, Jahromi ART (2012) Electrochemical performance of lead acid battery using ammonium hydrogen sulphate with different alkyl groups. Ionics 18:109–116
Lota G, Baraniak M, Wasiński K, Jankowska E (2013) Use of the HSO4 - groups-containing ionic liquids in lead-acid cells. Przem Chem 92:1624–1626
Rezaei B, Mallakpour S, Taki M (2009) Application of ionic liquids as an electrolyte additive on the electrochemical behavior of lead-acid battery. J Power Sources 187:605–612
Sato T, Masuda G, Takagi K (2004) Electrochemical properties of novel ionic liquids for electric double layer capacitor applications. Electrochim Acta 49:3603–3611
Lewandowski A, Świderska-Mocek A (2009) Ionic liquids as electrolytes for Li-ion batteries – an overview of electrochemical studies. J Power Sources 194:601–609
Stasiewicz M, Materna K, Pęziak-Kowalska D, Lota G (2015) Posibility of using 1-alkoxymethyl-4-(dimethylamine)pyridinium bis(trifluoromethylsulfonyl)imides as electrolytes for electrochemical capacitors. Int J Electrochem Sci 10:10513–10523
Zhang QB, Hua YX (2009) Corrosion inhibition of mild steel by alkylimidazolium ionic liquids in hydrochloric acid. Electrochim Acta 54:1881–1887
Zhang QB, Hua YX (2010) Corrosion inhibition of aluminum in hydrochloric acid solution by alkylimidazolium ionic liquids. Mater Chem Phys 119:57–64
Torriero AAJ (2015) Electrochemistry in ionic liquids vol. 1: fundamentals. Springer, Cham
Von Zamory J, Bedu M, Fantini S, Passerini S, Paillard E (2013) Polymeric ionic liquid nanoparticles as binder for composite Li-ion electrodes. J Power Sources 240:745–752
Funding
The authors would like to acknowledge gratefully the financial support from the National Centre of Research and Development of Poland, grant no. PBS3/A5/43/2015.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Grzegorz Lota is a ISE member
Electronic supplementary material
ESM 1
(DOCX 380 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gabryelczyk, A., Kopczyński, K., Baraniak, M. et al. Electrochemical properties of positive electrode in lead-acid battery modified by ammonium-based ionic liquids. J Solid State Electrochem 22, 919–930 (2018). https://doi.org/10.1007/s10008-017-3817-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-017-3817-7