
Abstract
The activated carbons (ACs) are prepared from mesophase pitch which is pre-carbonized by using potassium hydroxide as an activating agent. Nitrogen adsorption at 77 K is used to characterize the surface area and the porous structure of the ACs. By changing the pre-carbonization temperature from 560 to 750 °C, the arrangement of the ACs’ microcrystallines might be controllable, and the pore size could be adjusted between 1.5 and 2.4 nm. The electrochemical performance of the ACs in organic solvent (1 M Et4NBF4 in propylene carbonate) is investigated by voltage sweep cyclic voltammetry and constant current charge–discharge cycling tests. The results show that the ions can transfer rapidly and freely in the pore larger than or equal to 1.85 nm, even when the current density increases to 25 A g−1.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
As an energy storage unit, electric double-layer capacitors (EDLCs) [1, 2] have been studied and applied widely. It can store much more energy than conventional capacitors and offer much higher power density than batteries. Carbon materials are the main electrode materials which are based on the electrostatic interactions determining good reversibility and cyclability [3–5]. Activated carbons [6, 7], carbon aerogels [8], and carbon nanotubes [9] have been investigated in details.
The capacitance of EDLCs depends not only on the surface area of carbon materials but also on the pore size distribution (PSD) [1, 10–13]. Several studies reported an important capacitive contribution from micropores (<2 nm) [14, 15]; however, the micropores may be inaccessible for the solvated ions, so that the total surface area cannot be utilized fully. The mesopores are considered to be one of the key factors determining the capacitive performance of EDLCs because the ions can move freely in such pores. Thus, much attention has been attracted about preparing carbon materials with different ratios of micropore/mesopore and adjusting the PSD. Template carbonization technique [16–18] may adjust the mesopore size distribution by choosing different templates (such as zeolite, magnesium oxide, and silica) and changing the ratio of the template and carbon precursor; however, template carbonization technique increases carbon materials’ cost. Carbide-derived carbons (CDC) [11, 19] possess highly tailorable porosity, but the preparation cannot be scaled up in a cost-effective way.
Activated carbons are the main electrode materials for EDLCs due to their relatively low cost, simple process, and high surface area [6, 7, 13, 20, 21]. The PSD strongly depends on the types of their precursors and activation processes [13, 22]; however, the PSD of activated carbons (ACs) is more difficult to adjust, comparing with template carbons and CDC. Carbonization process has a remarked effect on the microstructure of carbon materials [23–28]. Wang [23, 25] investigated that petroleum coke-based and potato starch-based ACs were used as electrode materials for electrochemical capacitors, and concluded that the surface area and the volume of micropores and mesopores decreased with increasing pre-carbonization temperature. Raymundo-Pinero [24] studied nanoporous texture of activated carbons, and founded that pre-carbonization reduced mesopore volume between 1.6 and 3.2 nm and the pore distribution smaller than 0.7 nm did not change. It is well known that activated carbons are composed of disorderly graphitic microcrystallines which form different pores and create the surface area [28]. Pre-carbonization before activation process promotes the growth and orderly arrangement of graphitic microcrystallines, hinders the crystallites from intersecting with each other, and reduces the surface area of ACs [29, 30]. Thus, combining the pre-carbonization with the activation process, the stacking of ACs’ microcrystallines may be controllable, and the PSD can be adjusted. It is not reported that pre-carbonization process is considered as a method controlling the PSD effectively.
In our previous work [30], the activated carbons were prepared from mesophase pitch with KOH etching. The AC activated at 800 °C for 1 h with the KOH/MP ratio of 4:1 had a capacitance as high as 145 F/g. By changing the residence time at 800 °C, the intersection of the ACs’ microcrystallines was adjusted, and the pore size varied between 1.7 and 2.1 nm. In this report, we will focus on adjusting the PSD of ACs intensively by combining the pre-carbonization with activation process, and the mechanism of adjusting the PSD will be also studied in view of microstructure point. The obtained ACs are used to fabricate electrodes of EDLCs, and their electrochemical properties are measured.
Experimental
Preparation of activated carbons
Mesophase pitch (MP) with a softening point of 290 °C, supplied by Mitsubishi Gas Chemical Co., was used as the precursor of activated carbons. The pre-carbonization process was described as follows: MP was placed in stainless steel vessel heated in an argon flow. The carbonization temperature ramp was carried out at a rate of 2 °C min−1and held at different temperatures for 1 h. The temperature varied from 560–750 °C. Then, the carbonized MP was activated according to the following process—The carbonized MP and KOH solution were mixed, dried at 90 °C in vacuum oven, and placed into stainless steel vessel with the weight ratios of 1:4 and 1:5. The activation temperature ramp was carried out at a rate of 2 °C min−1, held at 800 °C for 1 h, and cooled in an argon flow. The obtained mixture was neutralized with HCl solution, filtered, and washed with distilled water until pH value reached 6–7. Finally, prepared activated carbons were dried at 85 °C for 48 h, followed by grinding and sieving to particle size less than 40 μm. The samples were labeled as MPy/xxx (y represents the mass ratio of KOH/carbonized MP and xxx represents carbonization temperature).
Characterization
The specific surface area and pore volume of the ACs were determined by nitrogen gas adsorption at 77 K. An automated adsorption apparatus (Micromeritics ASAP 2020) was employed for these measurements. The surface area was determined from the Brunauer–Emmett–Teller (B.E.T.). The total pore volume was determined from a point on the isotherm at a relative pressure above 0.98. The microporous volume was obtained using the t-plot method. The mesoporous volume was obtained by subtraction of the microporous volume from the total pore volume. The PSD was determined by density function theory (DFT) [31].
The powder X-ray diffraction (XRD) patterns of AC powder were obtained by using a D/MAX 2500 diffractometer (Rigaku Corporation, Japan) in which Cu-Kα (λ = 0.154 nm) was used as the radiation source.
Preparation of electrodes and a test capacitor
Activated carbon powder, acetylene black (as a conductive additive), and polyvinylidene fluoride (PVDF) were mixed at a mass ratio of 8:1:1 and dissolved in N-methyl pyrrolidone. The slurry was coated onto an aluminum (Al) foil (as a current collector). The carbon-coated Al foil was then dried under vacuum at 110 °C for 4 h, and punched in required size (1.5 cm2) as electrodes.
All the capacitors were assembled in a glove box, which had been vacuumed and then filled with argon. A sandwich-type capacitor was prepared with a pair of the carbon electrodes and the separator made from polypropylene. The 1 M Et4NBF4 in propylene carbonate was used as the electrolyte.
Electrochemical measurement
The capacitors were characterized by constant current charge—discharge and cyclic voltammograms (CVs) measurements from 0–2.5 V. All the tests were performed with a battery system VMP3 (Bio-Logic Corp., France). The specific discharge capacitance of the electrodes (C) in EDLCs can be calculated according to
where I is the discharge current, t is the discharge time, m is the activated carbon mass on an electrode, and △U is the voltage difference in discharge, excluding the portion of IR drop.
Results and discussion
Surface area and pore distribution of activated carbons
The characterization of the activated carbons derived from mesophase pitch is presented in Table 1. It shows that the surface area of ACs decreases drastically as the carbonized temperature increases. When the mass ratio (KOH/carbonized MP) is 4, the surface area decreases from 2,058–687 m2 g−1; when the ratio increases to 5, the surface area decreases from 2,583–750 m2 g−1.
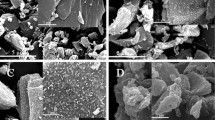
Carbonization promotes the growth of ACs’ microcrystallines [32] and makes crystallites inclined to orderly arrangement. Figure 1 illustrates the XRD patterns of ACs at the 4/1 ratio (KOH/carbonized MP). The XRD patterns of the ACs at the ratio of 5/1 are similar. There is a broad diffraction peak around 2θ = 25° in the patterns, corresponding to the diffraction of (002). As carbonized temperature rises, the intensity of peak (002) increases, suggesting that the three-dimensional arrangement is gradually ordered. Figure 2 [32] illustrates the effect of carbonization on the nanotexture of ACs. Graphitic microcrystallines intersect with each other and form different pores. With increasing heat temperature, the dimensions of crystallites get larger, and the arrangement of crystallites transfers from disorder to order status. When crystallites intersect disorderly, pores are formed easily; when the arrangement of crystallites is inclined to order status, pores got more difficultly in forming, and the surface area decreases. Thus, the higher the carbonized temperature is, the smaller the surface area of ACs is.

XRD patterns of ACs at the ratio of KOH/pitch (4/1)

Structural models ACs at different pre-carbonization temperatures
Figure 3 shows the variation of the PSD evaluated by DFT. The two dominant peaks are observed in all the curves. One area denoted as A locates from 0.5–1 nm, the other denoted as B appears from 1.5–2.5 nm. As carbonized temperature rises, the peak value decreases in B, as shown in Table 1. The peak value of ACs at the ratio of 4/1 decreases from 2.0–1.5 nm, and that at the ratio of 5/1 changes from 2.4–1.5 nm. The peak value in A does not change obviously with pre-carbonized temperature, which is in agreement with previous report [24]. From Fig. 2, increasing pre-carbonization temperature promotes the growth of crystallites and changes the arrangement of crystallites, and correspondingly the pores are changed. Thus, it indicates that the PSD of ACs can be adjusted by changing pre-carbonization temperature.

The PSD of ACs at different ratio of KOH/pitch. a At the ratio of KOH/pitch (4/1). b At the ratio of KOH/pitch (5/1)
Electrochemical characterization of activated carbons
Constant current charge–discharge tests were carried out between 0 and 2.5 V. Figure 4 shows the discharge curves of sample MP4/600 at different discharge currents. The other samples’ curves are similar. It can be seen that the voltage decreases linearly with the time, which indicates that carbon electrodes possess a better capacitive performance. The CVs were measured at the scan rate of 2 ∼ 50 mV s−1, as shown in Fig. 5. When the scan rate increases to 50 mV s−1, the CV curve still remains rectangular, which implies good rate performance.

Discharge curves at different discharge currents of the sample MP4/600

Cyclic voltammogram of the sample MP4/600 at different scan rates
The specific capacitance was calculated according to Eq. 1. Figure 6 shows the variation of the specific capacitance with the current density. From Fig. 6a, it can be seen that as the current density increases, the specific capacitance of samples MP4/560 and MP4/600 decreases a little and that of the other four samples decreases more and more drastically with increasing pre-carbonization temperature. In Fig. 6b, it is found that with increasing current density, the specific capacitance of samples pre-carbonized from 560–670 °C decreases a little and that of the other two samples decreases sharply. Referring to Table 1, the rule is obtained that when the peak value in zone B is larger than or equal to 1.85 nm, the capacitance of samples decreases a little with increasing current density and when the peak value is smaller than or equal to 1.75 nm, the capacitance of samples decreases drastically.

Variation of the specific capacitance with the current density. a ACs at the ratio of KOH/pitch (4/1). b ACs at the ratio of KOH/pitch (5/1)
It was suggested that the ideal size of the cation and anion solvated in 1 M Et4NBF4/PC was 1.96 and 1.71 nm, respectively [10, 33]. Besides, in organic electrolytes, it may occur that the ions are partially desolvated for entering the micropores [34]. Thus, the size of the cation and anion may get smaller. It is well known that the ions move more freely in larger pores and transfer more slowly in smaller pores [13]. When the ions transfer in the pores larger than the size of the ions, the ions may move very freely. As the current density increases, the ions can move more rapidly and form the electric double layer simultaneously at the electrode/electrolyte interface. Thus, the capacitance decreases a little. When the size of the pores is close to or slightly less than the ions, the ions may reduce their size by desolvation. When the size of the pores decreases sharply, the desolvated ions cannot move freely in smaller pores with increasing current density, and thus the amount of ions forming the electric double layer reduces, and the capacitance decreases drastically. From Table 1 and Fig. 6, it can be found that the capacitance of samples with the pore larger than or equal to 1.85 nm in zone B decreases a little with increasing current density and that the capacitance with the pore smaller than or equal to 1.75 nm decreases drastically. Thus, it suggests that the ions can transfer freely in the pore larger than or equal to 1.85 nm, even when the current density increases; on the contrary, the ions cannot move freely in the pore smaller than or equal to 1.75 nm, and the pore range of 1.75 ∼ 1.85 nm may be the critical one in 1 M Et4NBF4/PC.
The cyclability of samples MP4/600 and MP5/600 at a current density of 5 A g−1 is shown in Fig. 7. At 25 °C, the reduction of samples MP4/600 and MP5/600 in the initial specific capacitance is estimated in 8% and 10% after 10,000 cycles, respectively. When the measurement temperature rises to 60 °C, the reduction of two samples is in about 16% after long-term cycling. It indicates that the samples MP4/600 and MP5/600 have stable cyclability at large current density, under 25 and 60 °C.

Variation of the specific capacitance on the number of cycles at a current density of 5 A g−1 under 25 and 60 °C
The relation between the capacitance and pore size distribution
The micropore surface area and the mesopore surface area contribute to the capacitance together [6]. The following relation is established as follows:
where \( C_{\rm{dl}}^{\rm{mic}} \) and \( C_{\rm{dl}}^{\rm{mes}} \) are the capacitance per unit micropore surface area and the capacitance per unit mesopore surface area, respectively. S mic and S mes are the micropore surface area and mesopore surface area, respectively. We can get the following relation from Eq. 2:
In Fig. 8, C/S mes is plotted against S mic /S mes as the capacitance measured at the current density of 0.5 A g−1. It can be seen that the data is mainly fitted to the linear relation, except that a few dots deviate a little from the fitted line at top of Fig. 8.

C/Smes plots as a function of Smic/Smes for ACs at the current density of 0.5 A g−1
We can also get the following relation from Eq. 2:
Figure 9 shows the variation of the C/S mic as a function of S mes /S mic ratio for all the prepared ACs in this work. It can be seen that all the plots can be divided into two categories, and the data of each category is fitted in linear relationship. Category 1 includes six samples whose peaks in zone B are exactly larger than or equal to 1.85 nm, and the other six samples whose peaks are all smaller than or equal to 1.75 nm belong to category 2. The \( C_{\rm{dl}}^{\rm{mic}} \) (9.6 μF cm−2) of category 1 is larger than that (3.4 μF cm−2) of category 2, which indicates that the contribution of micropore surface area in category 1 is larger than that in category 2. As the values of \( C_{\rm{dl}}^{\rm{mic}} \) and \( C_{\rm{dl}}^{\rm{mes}} \) in Eq. 4 are calculated according to part of all the data, their values are different from those in Eq. 3.

C/Smic plots as a function of Smes/Smic for ACs at the current density of 0.5 A g−1
Equation 2 introduces the model which takes into the difference between the specific capacitance on micropore surface area and that on mesopore surface area. Equation 3, as one form of Eq. 2, has been applied widely [35–37]. On the basis of the form, the \( C_{\rm{dl}}^{\rm{mic}} \) is found to be close, and the \( C_{\rm{dl}}^{\rm{mes}} \) changes due to the mesoporous structure and surface morphology.
In organic electrolytes, the ions’ movement may be blocked in micropores, as the ions in organic electrolytes are larger than those in aqueous electrolytes [38]. In the present work, in 1 M Et4NBF4/PC, the ions can transfer freely in micropores larger than or equal to 1.85 nm and move slowly in those smaller than or equal to 1.75 nm. Thus, the capacitance per unit micropore surface area may be different. Equation 4, as another form of Eq. 2, may focus on the effect of the micropore surface area on the capacitance. In Fig. 9, the data is classified into two categories according to the size of micropore in area B, and each category is well fitted with linear relationship. As the pore larger than or equal to 1.85 nm makes the ions transfer freely, \( C_{\rm{dl}}^{\rm{mic}} \) of category 1 is larger than that of category 2.
The relationship between the capacitance and the surface area is also investigated at the current density of 10 A g−1, as shown in Figs. 10 and 11. Comparing Fig. 10 with Fig. 8, the value of \( C_{\rm{dl}}^{\rm{mes}} \) rises by 52% from 2.3–3.5 μF cm−2, and the \( C_{\rm{dl}}^{\rm{mic}} \) decreases by 17% from 12–10 μF cm−2. With increasing current density, the ions move more easily in the mesopores than in the micropores [6, 37]. Thus, the contribution of mesopore surface area gets larger, and that of micropore surface area decreases. The variation amplitude of \( C_{\rm{dl}}^{\rm{mes}} \) is larger than that of \( C_{\rm{dl}}^{\rm{mic}} \), which indicates that Eq. 3 is fit for analyzing the capacitance per unit mesopore surface area. In Fig. 11, \( C_{\rm{dl}}^{\rm{mic}} \) and \( C_{\rm{dl}}^{\rm{mes}} \) of category 1 are almost the same as those in Fig. 9. It indicates that the ions can move freely in pores of category 1 at large current density. For category 2, the value of \( C_{\rm{dl}}^{\rm{mic}} \) falls from 3.4–2.4 μF cm−2, and \( C_{\rm{dl}}^{\rm{mes}} \) increases a little. It can be seen that Eq. 4 may contribute to studying the contribution of the micropore area.

C/Smes plots as a function of Smic/Smes for ACs at the current density of 10 A g−1

C/Smic plots as a function of Smes/Smic for ACs at the current density of 10 A g−1
Conclusions
By changing pre-carbonization temperature from 560 to 750 °C, the arrangement of microcrystallines is controllable, and correspondingly the PSD of mesophase pitch-based ACs could be adjusted. The size of pore between 1.5 and 2.4 nm decreases with increasing pre-carbonization temperature. The electrochemical performance of ACs has been investigated in 1 M Et4NBF4/PC. It is found that the pore larger than or equal to 1.85 nm is in favor of ions’ movement, which favors to obtain a high capacitance with high current density; on the contrary, the ions cannot move freely in the pore smaller than or equal to 1.75 nm, and the pore range of 1.75 ∼ 1.85 nm may be the critical one in 1 M Et4NBF4/PC.
Equation 2 introduced the contribution of micropore surface area and mesopore surface area on the capacitance, respectively. Equation 3, as one form of Eq. 2, is helpful to analyze the capacitance per unit mesopore surface area. Equation 4, as another one, may focus on the effect of the micropore surface area on the capacitance.
References
Yang Y (2006) Battery 36:34
Kotz R, Carlen M (2000) Electrochim Acta 45:2483
Janes A, Permann L, Arulepp M, Lust E (2004) Electrochem Commun 6:313
Lust E, Nurk G, Janes A, Arulepp M, Nigu P, Moller P, Kallip S, Sammelselg V (2003) J Solid State Electrochem 7:91
Fang BZ, Binder L (2006) J Phys Chem B 110:7877
Shi H (1996) Electrochim Acta 41:1633
Qu DY, Shi H (1998) J Power Sources 74:99
Probstle H, Wiener M, Fricke J (2003) J Porous Mater 10:213
Pico F, Rojo JM, Sanjuan ML, Anson A, Benito AM, Callejas MA, Maser WK, Martinez MT (2004) J Electrochem Soc 151:A831
Endo M, Kim YJ, Ohta H, Ishii K, Inoue T, Hayashi T, Nishimura Y, Maeda T, Dresselhaus MS (2002) Carbon 40:2613
Largeot C, Portet C, Chmiola J, Taberna PL, Gogotsi Y, Simon P (2008) J Am Chem Soc 130:2730
Wu FC, Tseng RL, Hu CC, Wang CC (2005) J Power Sources 144:302
Pandolfo AG, Hollenkamp AF (2006) J Power Sources 157:11
Simon P, Gogotsi Y (2008) Nat Mater 7:845
Chmiola J, Yushin G, Gogotsi Y, Portet C, Simon P, Taberna PL (2006) Science 313:1760
Jurewicz K, Vix-Guterl C, Frackowiak E, Saadallah S, Reda A, Parmentier J, Patarin J, Beguin F (2004) J Phys Chem Solids 65:287
Sevilla M, Alvarez S, Centeno TA, Fuertes AB, Stoeckli F (2007) Electrochim Acta 52:3207
Fuertes AB, Lota G, Centeno TA, Frackowiak E (2005) Electrochim Acta 50:2799
Chmiola J, Yushin G, Dash R (2006) J Power Sources 158:765
Zheng JP, Huang J, Jow TR (1997) J Electrochem Soc 144:2026
Probstle H, Schmitt C, Fricke J (2002) J Power Sources 105:189
Kim YJ, Lee BJ, Suezaki H, Chino T, Abe Y, Yanagiura T, Park KC, Endo M (2006) Carbon 44:1592
Zhao S, Wang CY, Chen MM, Wang J, Shi ZQ (2009) J Phys Chem Solids 70:1256
Raymundo-Pinero E, Kierzek K, Machnikowski J, Beguin F (2006) Carbon 44:2498
Shi ZQ, Zhao S, Chen MM, Wang MX, Wang CY (2008) J Inorg Mater 23:799
Tian Y, Song Y, Tang ZH, Guo QG, Liu L (2008) J Power Sources 184:675
Ruiz V, Blanco C, Raymundo-Pinero E, Khomenko V, Beguin F, Santamaria R (2007) Electrochim Acta 52:4969
Lu CL, Xu SP, Gan YX, Liu SQ, Liu CH (2005) Carbon 43:2295
Weng TC, Teng HS (2001) J Electrochem Soc 148:A368
Zhai D, Li B, Kang F (2010) Microporous Mesoporous Mater 130:224
Seaton NA, Walton JPRB, Quirke N (1989) Carbon 27:853
Inagak M, Kang FY (2006) Carbon materials science and engineering-From fundamentals to applications. Tsinghua University Press, Beijing
Kim YJ, Masutzawa Y, Ozaki S, Endo M, Dresselhaus MS (2004) J Electrochem Soc 151:E199
Lin R, Taberna PL, Chmiola J, Guay D, Gogotsi Y, Simon P (2009) J Electrochem Soc 156:A7
Gryglewicz G, Machnikowski J, Lorenc-Grabowska E (2005) Electrochim Acta 50:1197
Wang LH, Toyoda M, Inagaki M (2008) New Carbon Mater 23:111
Wang LH, Fujita M, Inagaki M (2006) Electrochim Acta 51:4096
Hulicova D, Kodama M, Hatori H (2006) Chem Mater 18:2318
Acknowledgements
This work was financially supported by the Natural Science Foundation of China under grant no. 50632040 and no. 50802049. We also appreciate the financial support from Guangdong Province Innovation R&D Team Plan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhai, D., Li, B., Du, H. et al. The effect of pre-carbonization of mesophase pitch-based activated carbons on their electrochemical performance for electric double-layer capacitors. J Solid State Electrochem 15, 787–794 (2011). https://doi.org/10.1007/s10008-010-1156-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-010-1156-z