
Abstract
Context
The activation of C-H bonds is a fundamental process in synthetic organic chemistry, which enables their replacement by highly reactive functional groups. Coordination compounds serve as effective catalysts for this purpose, as they facilitate chemical transformations by interacting with C-H bonds. A comprehensive understanding of the mechanism of activation of this type of bond lays the foundation for the development of efficient protocols for cross-coupling reactions. We explored the activation of C(sp2)-H bonds in 1-Phenyl-4-vinyl-1H-1,2,3-triazole derivatives with CH3, OCH3, and NO2 substituents in the para position of the phenyl ring, using palladium acetate as catalyst. The studied reaction is the first step for subsequent conjugation of the triazoles with naphthoquinones in a Heck-type reaction to create a C–C bond. The basic nitrogen atoms of the 1,2,3-triazole coordinate preferentially with the cationic palladium center to form an activated species. A concerted proton transfer from the terminal vinyl carbon to one of the acetate ligands with low activation energy is the main step for the C(sp2)-H activation. This study offers significant mechanistic insights for enhancing the effectiveness of C(sp2)-H activation protocols in organic synthesis.
Methods
All calculations were performed using the Gaussian 09 software package and density functional theory (DFT). The structures of all reaction path components were fully optimized using the CAM-B3LYP functional with the Def2-SVP basis set. The optimized geometries were analyzed by computing the second-order Hessian matrix to confirm that the corresponding minimum or transition state was located. To account for solvent effects, the Polarizable Continuum Model of the Integral Equation Formalism (IEFPCM) with water as the solvent was used.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The activation of C(sp2)-H bonds is a crucial process in several fields of organic synthesis, as this transformation allows the replacement of a C(sp2)-H bond by a new bond, resulting in a highly reactive functionalized carbon atom [1, 2]. Transition metal complexes have been extensively employed to activate C(sp2)-H bonds, leading to the development of various synthetic methodologies. The availability of abundant carbon-rich biomass has renewed interest in C(sp2)-H bond activation as a first step towards the synthesis of fuels and fine chemicals, as highlighted by several studies [3,4,5,6].
Coordination compounds act as effective catalysts for C(sp2)-H activation by direct interaction of the C-H bond with the metallic center, allowing for activation and subsequent functionalization. This interaction provides a practical alternative for achieving chemical transformations, as the activated C-H bonds become a center of high reactivity [7]. This offers a viable alternative for achieving the desired chemical transformations. Understanding the mechanism and kinetic parameters for such activation is essential towards creating more effective functionalization protocols [8,9,10,11,12,13].
The field of catalytic activation of C-H bonds has seen significant advances in recent years. Since the description of the acetolysis reaction of diphenyl mercury in acetic acid was first proposed by Winstein and Traylor in 1955, the work of Fagnou et al. has presented crucial aspects for the understanding of the mechanisms of C-H bond activation [14]. The use of transition metal catalysts in the direct coupling reaction is a useful tool in the regio- and chemo-selective control to obtain hetero arenes. These studies collectively underscore the growing importance and potential of catalytic activations not only of C-H bonds but also of C-O bonds in organic synthesis [15, 16].
C-H bond activation protocols typically involve the coordination of a metal to a hydrogen atom, followed by a reaction step that forms a new product. The choice of metal, ligands, and reaction conditions can have a significant impact on the efficiency and selectivity of the reaction. In this regard, palladium-based catalysts have been widely used due to their ability to activate C-H bonds on a variety of substrates. In addition, several studies have pointed to the use of auxiliary ligands that have allowed the selective activation of C-H bonds in complex molecules [17, 18].
In this context, the mechanism for activation of C-H bonds can occur by a variety of pathways, such as concerted metalation deprotonation (CMD), aromatic electrophilic substitution (SeAr), internal electrophilic substitution (IES), and base-assisted IES (BIES) and ambiphilic metal-ligand assistance (AMLA), [19,20,21,22,23,24] thus broadening the scope and potential of this field of research on C-H activation mechanisms [25].
Computational methods are a valuable tool for exploring the inherent complexity of the carboxylate-assisted C-H bond activation process. This process is well understood and can be confidently applied in various contexts [26,27,28]. Studies dedicated to characterizing C-H-assisted cleavage with various metals have shown that the interaction between the C-H bond and an electron-deficient metal center makes the bond susceptible to cleavage by a chelating carboxylate base [29, 30].
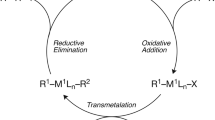
The goal of the present study is to examine the activation parameters for the initial step of the reaction between 1-Phenyl-4-vinyl-1H-1,2,3-triazole derivatives and 1,4-naphthoquinones, catalyzed by Pd(OAc)2, in a Heck-type reaction to form a new C-C bond. In the course of our investigation, we observed that the 1,2,3-triazoles in the presence of the Pd(OAc)2 catalyst lead to a complex which allows for C(sp2)-H activation by a concerted proton transfer from the terminal vinyl carbon to one of the acetate groups of the catalyst. Additionally, we also investigate the effect of para substituents in the phenyl ring on the activation parameters, as it has been shown that these substituents may induce changes in the reaction product after coupling with the naphthoquinones. Scheme 1 illustrates the catalytic cycle of the reaction between the 1-Phenyl-4-vinyl-1H-1,2,3-triazole derivatives and 1,4-naphthoquinone derivatives mediated by Pd(OAc)2 in a Heck-type reaction culminating in the formation of a new C-C bond [31]. In the present study, we concentrate on the first step of the reaction, coordination of the 1,2,3-triazole to the Pd(OAc)2 catalyst followed by activation of the C(sp2)-H bond, which will finally lead to the formation of a new C-C bond after coordination with the naphthoquinone.

a Reaction conditions for the synthesis of product 6. b Representation of the proposed catalytic cycle for the reaction between 1-Phenyl-4-vinyl-1H-1,2,3-triazol and 1,4-naftoquinone mediated by a Pd(OAc)2 catalyst. Coordination mode A is the coordination via the N-3 atom of the 1,2,3-triazole ring (4a) and coordination mode B is coordination via the C = C double bond of the vinyl moiety (4b) [31]
Computational methods
All calculations were performed using the Gaussian 09 software package, with computations using the density functional theory [32]. All structures involved in the reaction path (reactants, products, intermediates, and transition states) were fully optimized using the CAM-B3LYP functional [33] with the Def2-SVP basis set [34]. For the optimized geometries, we computed the second order Hessian matrix to confirm them as a minimum energy structure (no negative eigenvalue) or as a transition state (just one negative eigenvalue). Solvent effects were included through the implicit solvation model using the Polarizable Continuum Model of the Integral Equation Formalism (IEFPCM), with water as solvent (ε = 78.35) [35]. Relative energies were computed in kcal mol−1, taking the isolated reactants (1-Phenyl-4-vinyl-1,2,3-triazole derivatives and the Pd(OAc)2 complex) as reference. The normal vibrational modes calculations enabled us to obtain thermodynamic and kinetic parameters (at T = 298 K and p = 1 atm) by employing the standard statistical thermodynamic equations for an ideal gas [36]. Energy values in the text are discussed in terms of ΔH (kcal mol−1). The coordinates of all structures discussed in this text, including transition structures, are given in the Supporting Information.
Results and discussion
General aspects
Our first step was to investigate the coordination mode of the 1-Phenyl-4-vinyl-1,2,3-triazole to the Pd(OAc)2 complex. Previous studies on this first step considered only the coordination via the C=C double bond of the vinyl moiety [31]. However, when we tried the alternative coordination via the basic nitrogen atoms of the 1,2,3-triazole ring, we found that the pre-reactive complex (4a) is 8.98 kcal mol−1 more stable than coordination via the C=C double bond of the vinyl moiety (4b) (Scheme 2; all energy values discussed in this section are for the R = H derivative). Following the relative basicity order, N-3 of the 1,2,3-triazole ring coordinates preferentially to the metal center [37, 38]. The two alternative pre-reactive complexes and their relative energies are given in Scheme 2.

Energy profiles for the initial steps of the coupling reaction between the 1-Phenyl-4-vinyl-1,2,3-triazole derivatives and the Pd(OAc)2 complex. The energy data are for the R = H derivative. The grey line corresponds to the coordination mode B, with the metal center coordinating via the C = C double bond. The orange line corresponds to the coordination mode A, with the metal center coordinating via the N-3 atom of the 1,2,3-triazole ring. The blue line represents the alternative path after coordination via the C = C double bond
In both cases, one of the acetate groups is partially uncoordinated, so that the Pd atom maintains the planar tetra coordination mode and the +2 oxidation state. In the structure coordinated by the N-3 atom (4a), the C=C double bond is facing the Pd cation, with one of the hydrogens of the terminal carbon pointing towards the uncoordinated oxygen atom of the acetate group, a point that is relevant for the next step. Differentley, in the structure coordinated by the C=C double bond (4b), the triazole ring is far apart from the Pd center.
We also computed the transition structures leading to these initial complexes. Their relative energies follow the same trend of the relative energies of the stable complexes. Activation energy to coordination via the N-3 atom of the triazole ring (4b-TS) is 4.40 kcal mol−1 (Scheme 2), while activation energy to coordination via the C=C double bond (4b-TS) is 9.72 kcal mol−1 (Scheme 2).
Starting from the less stable initial complex (4b), the rotation of the 1,2,3-triazole ring may bring it to an orientation where it can undergo a nucleophilic attack of the oxygen atom of the uncoordinated acetate group on the C=C double bond of the vinyl moiety. Both carbon atoms of the C=C double bond could undergo the nucleophilic attack to form an intermediate bicyclic complex. Although the complex formed upon attack on the terminal carbon (5b*) is less stable than the one formed upon attack on the carbon atom vicinal to the triazole ring (5b) by 5.65 kcal mol−1 (Scheme 2), the activation energy to form 5b (5b-TS) is smaller than that to form 5b* (5b*-TS) by 5.15 kcal mol−1 (Scheme 2). Additionally, we could not envisage any pathway further from the 5b* structure. In contrast, starting from 5b-TS, a second displacement of the acetate group, promoted by the N-3 of the 1,2,3-triazole ring, leads finally to the bicyclic complex 5b. This is the most stable species along this pathway (Scheme 2). Therefore, by coordination of the 1-Phenyl-4-vinyl-1,2,3-triazole derivatives to the Pd(OAc)2 complex via the C=C double bond, the final species is the bicyclic complex 6b, with one of the acetate group bonded to the carbon atom vicinal to the triazole ring. Observe that, in this case, there is a rehybridization of the carbon atoms of the vinyl group, going from the original sp2 hybridization to the sp3 hybridization seen in the bicyclic species 6b. The new bonds required for this rehybridization are formed one with the Pd center and another one with the uncoordinated acetate group. 6b is 18.26 kcal mol−1 more stable than the starting reactants (Scheme 2). Along this path, the highest energy structure is the transition state 5b-TS, with a relative energy of 9.72 kcal mol−1 (Scheme 2).
A completely different picture emerges when considering the alternative coordination mode, via the N-3 atom of the 1,2,3-triazole ring. Firstly, the initial complex (4a) is more stable than the corresponding 4b by 8.98 kcal mol−1 (Scheme 2). Secondly, as we will show below, the highest energy point on this pathway also has lower relative energy than the highest energy point on the pathway described before.
As pointed out before, after coordination of the 1-Phenyl-4-vinyl-1,2,3-triazole derivatives to the Pd(OAc)2 complex via the N-3 atom (4a), the C = C double bond is found facing the Pd atom, with one of the hydrogen atoms of the terminal carbon of the vinyl group pointing towards the uncoordinated oxygen atom of the acetate group. In an attempt to find a pathway going ahead from 4a, we arrived at a highly stable structure (5a), with correspondingly low activation energy to form it (5.02 kcal mol−1, Scheme 2), resulting from a proton transfer from the terminal carbon atom of the vinyl group to the uncoordinated oxygen atom of the acetate group and formation of a C-Pd bond, in a concerted process. Processes similar to this one have been largely reported before in connection with the activation of the C(sp2)-H bond, catalyzed by transition metal complexes, like the one employed in the present study [7, 39,40,41]. Notably, the activation energy required for this concerted step is lower (by 5.02 kcal mol−1) than that for the alternative route, starting with the coordination of the double bond to the palladium center.
The final structure in this alternative path is also a bicyclic species where the N-3 nitrogen atom of the 1,2,3-triazole ring and the carbon atom of the vinyl group are both coordinated to the palladium atom. One of the acetate groups completely detaches from the palladium center in the form of an acetic acid molecule. In this case, there is no rehybridization of the carbon atoms of the vinyl group, which maintain their C(sp2) nature. Observe that, in both pathways, the palladium center maintains the formal +2 oxidation state.
With the results discussed above, we are proposing that, upon coordination of a 1-Phenyl-4-vinyl-1,2,3-triazole structure to the palladium diacetate, the preferential coordination mode is via the basic N-3 atom of the 1,2,3-triazole ring leading to a bicyclic species where both the N-3 atom and the terminal carbon of the vinyl group are coordinated to the palladium atom. The highest energy point on this pathway is the transition structure 5a-TS, with a relative energy of 5.02 kcal mol−1 (Scheme 2). Therefore, the pathway via coordination of the N-3 atom of the 1,2,3-triazole followed by reaction with the vinyl group, goes through a transition structure having much lower energy, therefore being a faster process.
The analysis of interatomic distances around the metal center unequivocally demonstrates the formation of a C-Pd bond during the reaction in both pathways. The interatomic C-Pd distances in 5a and 6b are of 1.95 Å (C(sp2)-Pd) and 1.98 Å (C(sp3)-Pd), respectively, providing evidence for the coordination of the substrate to the metal center.
The thermodynamic feasibility of the reaction is indicated by the strong exothermicity in both pathways (−14.87 kcal.mol−1 for 5a and −18.26 kcal mol−1 for 6b (Scheme 2), with a much lower activation energy for the coordination mode A, via the N-3 atom of the 1,2,3-triazole ring (structure 5a).
The concerted metalation deprotonation (CMD) mechanism in the coordination mode A is supported by the fact that it allows for the simultaneous formation of the Pd-C bond, deprotonation of the substrate, and formation of the O-H bond, as shown in Fig. 1. The obtained data is consistent with this coordinated set of events, reinforcing the conclusion that the reaction occurs via the CMD mechanism [42,43,44,45,46].

Transition state snapshots and IRC calculation (side view) of acetate ligand-assisted intramolecular activation and deprotonation
The IRC coordinate reveals a subtle shift in the charge to the metallic center, hinting at a reductive transformation. This implies a rise in electron density on the metal center aligning with the expected behavior of a nucleophile. Such a change is indicative of a nucleophilic action, contributing to the electron density increase and suggesting a reductive process.
Substituent effects
Experimental evidence shows that substituents in the para position of the phenyl ring of the 1-Phenyl-4-vinyl-1,2,3-triazole derivatives may have some effect on the course of the reaction [31]. For the unsubstituted species (R = H), the yield of the reaction after coupling with the 1,4-naphtoquinone is 38%. Substitution by a methyl group (R = CH3) increases the reaction yield to 70%; substitution by a nitro group (R = NO2) reduces the yield to 28%, while substitution by a methoxy group (R = OCH3) leads to no reaction [31]. Computation of the relevant points along the reaction coordinate using the energy span protocol revealed that the relative stabilities and activation enthalpies of the main intermediates for the substituted derivatives do not significantly change as compared to the unsubstituted system, attributing the observed effects on the reaction yield to the experimental conditions [31]. We therefore investigated the effect of the given substituents on the thermodynamic and kinetic parameters for the two pathways we found. The main data for the coordination mode A are given in Table 1. The corresponding values for the coordination mode B are given in the Supporting Information.
The data in Table 1 reinforces the previous findings. Although there is some effect of the different substituents on the relative energies, they are not enough to drastically change the kinetic and thermodynamic parameters for the reaction, as compared to the unsubstituted species, in particular for R = CH3. The electron-releasing OCH3 group reduces the activation energy and increases the stability of the final product by 2.90 kcal mol−1, while the electron-withdrawing group NO2 increases the activation energy by 2.31 kcal mol−1 and reduces the stability of the final product by 2.15 kcal mol−1.
To rationalize the electronic effects of the substituents in the 1,2,3-triazole ring, we also analyzed their HOMO, and the results indicate a complex relation between the molecular structure and the electronic properties. The substitution seems to affect the electron density of the aromatic ring, consistent with organic chemistry’s understanding of the effect of substituents on conjugate systems (Fig. 2). However, the N1-N2-C3-C4 dihedral angle creates a torsion between the aromatic ring and the triazole group, disturbing the conjugation and limiting the extent of the electron delocalization (Fig. 2). This may explain the minimal variation observed in the electron density on the vinyl double bond. In addition, the small changes in the HOMO orbital energies for the substituents suggest that chemical reactivity may not be drastically affected by the nature of the substituent. The relative energies of the HOMOs, along with the disruption of the conjugation, may be the key to understanding the small effect on the energy of the transition states and final products. Thus, the substituent groups do not have a significant influence on the electron density of the double bond of the vinyl group and in the 1,2,3-triazole ring (Fig. 2).

Representation of HOMO orbitals, their energies, and the N1-N2-C3-C4 dihedral angles in the triazole derivatives
In any way, it is seen that electron-releasing substituents reduce the activation energy and stabilize the final product, while electron-withdrawing groups increase the activation energy and reduce the stability of the final product, although to a small amount.
Conclusion
In the present study, we investigated the coupling between 1-Phenyl-4-vinyl-1,2,3-triazole derivatives and palladium acetate. Our investigation show that the 1-Phenyl-4-vinyl-1,2,3-triazole derivatives preferentially coordinate to the Pd(OAc)2 complex via the N-3 atom of the 1,2,3-triazole ring, with low activation energy. After a proton transfer from the terminal carbon atom of the vinyl group to one of the acetate groups, with the formation of a C(sp2)-Pd bond and an acetic acid molecule, a highly stable intermediate is formed. Although the alternative pathway, with coordination of the 1-Phenyl-4-vinyl-1,2,3-triazole derivative to the palladium center via the C = C double bond of the vinyl group also leads to a highly stable intermediate, the activation energy for this path is much higher. Substituent effects are not so high, although electron-releasing substituents decrease the activation energy and increase the stability of the final intermediate, while the opposite is observed for the electron-withdrawing groups, which increase the activation energies and reduce the relative stability of the final intermediate.
Data availability
This published article and its supplementary information fle include all the data generated or analyzed during this study.
References
Labinger JA, Bercaw JE (2002) Understanding and exploiting C-H bond activation. Nature 417(6888):507–514. https://doi.org/10.1038/417507a
Dhanush PC, Saranya PV, Anilkumar G (2022) Microwave assisted C-H activation reaction: an overview. Tetrahedron 105:132614. https://doi.org/10.1016/J.TET.2021.132614
Moghimi S, Mahdavi M, Shafiee A, Foroumadi A (2016) Transition-metal-catalyzed acyloxylation: activation of C(sp2)–H and C(sp3)–H bonds. Eur J Org Chem 20:3282–3299. https://doi.org/10.1002/EJOC.201600138
Tzouras NV, Stamatopoulos IK, Papastavrou AT, Liori AA, Vougioukalakis GC (2017) Sustainable metal catalysis in CH activation. Coord Chem Rev 343:25–138. https://doi.org/10.1016/J.CCR.2017.04.012
Sahoo SR, Dutta S, Al-Thabaiti SA, Mokhtar M, Maiti D (2021) Transition metal catalyzed C-H bond activation by exo-metallacycle intermediates. Chem Commun 57(90):11885–11903. https://doi.org/10.1039/D1CC05042G
Landaeta VR, Rodríguez-Lugo RE (2017) Insights on the CH bond activation by transition metal complexes from groups 8–10 bearing (P-N) chelates. J Mol Catal A: Chem 426:316–325. https://doi.org/10.1016/J.MOLCATA.2016.08.011
Wei WX, Li Y, Wen YT, Li M, Li XS, Wang CT, Liu HC, Xia Y, Zhang BS, Jiao RQ, Liang YM (2021) Experimental and computational studies of palladium-catalyzed spirocyclization via a Narasaka–Heck/C(sp3 or sp2)–H activation cascade reaction. J Am Chem Soc 143(20):7868–7875. https://doi.org/10.1021/jacs.1c04114
Chang W, Wang Y, Chen Y, Ma J, Liang Y (2023) Control of meta-selectivity in the Ir-catalyzed aromatic C-H borylation directed by hydrogen bond interaction: a combined computational and experimental study. Chin Chem Lett 34(5):107879. https://doi.org/10.1016/j.cclet.2022.107879
Haag B, Mosrin M, Ila H, Malakhov V, Knochel P (2011) Regio- and chemoselective metalation of arenes and heteroarenes using hindered metal amide bases. Angew Chem Int Ed 50(42):9794–9824. https://doi.org/10.1002/anie.201101960
Vilhena FS, Bickelhaupt FM (2017) Carneiro JWM (2017) Regio- and stereoselectivity in 1,3-dipolar cycloadditions: activation strain analyses for reactions of hydrazoic acid with substituted alkenes. Eur J Org Chem 29:4313–4318. https://doi.org/10.1002/ejoc.201700577
Gorelsky SI (2013) Origins of regioselectivity of the palladium-catalyzed (aromatic) CH bond metalation-deprotonation. Coord Chem Rev 257(1):153–164. https://doi.org/10.1016/j.ccr.2012.06.016
Li M, Li SX, Chen DP, Gao F, Qiu YF, wamg XC, Quan ZS, Liang YM (2023) Regioselective C–H active carbonylation via 1,4-palladium migration. Organic Letters. https://doi.org/10.1021/ACS.ORGLETT.3C00567
Mandal A, Bera R, Baidya M (2021) Regioselective C-H alkenylation and unsymmetrical bis-olefination of heteroarene carboxylic acids with ruthenium catalysis in water. J Org Chem 86(1):62–73. https://doi.org/10.1021/acs.joc.0c02215
Lapointe D, Fagnou K (2010) Overview of the mechanistic work on the concerted metallation-deprotonation pathway. Chem Lett 39(11):1118–1126. https://doi.org/10.1246/cl.2010.1118
Boit TB, Bulger AS, Dander JE, Garg NK (2020) Activation of C-O and C-N bonds using non-precious-metal catalysis. ACS Catal 10(20):12109–12126. https://doi.org/10.1021/acscatal.0c03334
Villo P, Shatskiy A, Kärkäs MD, Lundberg H (2023) Electrosynthetic C−O bond activation in alcohols and alcohol derivatives. Angew Chem Int Ed 62(4). https://doi.org/10.1002/anie.202211952
Gorelsky SI, Lapointe D, Fagnou K (2008) Analysis of the concerted metalation-deprotonation mechanism in palladium-catalyzed direct arylation across a broad range of aromatic substrates. J Am Chem Soc 130(33):10848–10849. https://doi.org/10.1021/ja802533u
Rousseaux S, Gorelsky SI, Chung BKW, Fagnou K (2010) Investigation of the mechanism of C(sp3)-H bond cleavage in Pd(0)-catalyzed intramolecular alkane arylation adjacent to amides and sulfonamides. J Am Chem Soc 132(31):10692–10705. https://doi.org/10.1021/ja103081n
He F, Gourlaouen C, Pang H, Braunstein P (2022) Influence of the flexibility of nickel PCP-pincer complexes on C-H and P-C bond activation and ethylene reactivity: a combined experimental and theoretical investigation. Chemistry. https://doi.org/10.1002/chem.202104234
Ren Y, Yang T, Lin Z (2022) Insights into the mechanism and selectivity of the Rh(I)-catalyzed cycloisomerization reaction of benzylallene-alkynes involving C-H bond activation. Organic Chemistry Frontiers 10(1):115–126. https://doi.org/10.1039/d2qo01503j
Muhr M et al. (2022) C-H and Si-H activation reactions at Ru/Ga complexes: a combined experimental and theoretical case study on the Ru-Ga bond. Chemistry, 28(54). https://doi.org/10.1002/CHEM.202200887
Huang G, Fang Y, Wright JS, Ni SF, De Li M, Dang L (2023) The essence in selectivity of copper-mediated intermolecular nucleophilic substitution of a meta C-H bond in 2-Methyl- N-methoxyaniline: a theoretical study. J Phys Chem A 127(45):9473–9482. https://doi.org/10.1021/ACS.JPCA.3C05223
You F, Zeng J, Rouf AM, Dong S, Zhu J (2022) Theoretical study on reaction mechanisms of dinitrogen activation and coupling by carbene-stabilized borylenes in comparison with intramolecular C-H bond activation. Chem Asian J 17(12). https://doi.org/10.1002/ASIA.202200232
Sajjad MA, Harrison JA, Nielson AJ, Schwerdtfeger P (2018) NBO orbital interaction analysis for the ambiphilic metal–ligand activation/concerted metalation deprotonation (AMLA/CMD) mechanism involved in the cyclopalladation reaction of N N dimethylbenzylamine with palladium acetate. Organometallics 37(21):3659–3669. https://doi.org/10.1021/ACS.ORGANOMET.8B00303
Roudesly F, Oble J, Poli G (2017) Metal-catalyzed C-H activation/functionalization: the fundamentals. J Mol Catal A: Chem 426:275–296. https://doi.org/10.1016/j.molcata.2016.06.020
Ackermann L (2011) Carboxylate-assisted transition-metal-catalyzed C-H bond functionalizations: mechanism and scope. Chem Rev 111(3):1315–1345. https://doi.org/10.1021/cr100412j
Alharis RA, McMullin CL, Davies DL, Singh K, Macgregor SA (2019) The importance of kinetic and thermodynamic control when assessing mechanisms of carboxylate-assisted C-H activation. J Am Chem Soc 141(22):8896–8906. https://doi.org/10.1021/jacs.9b02073
Alharis RA, McMullin CL, Davies DL, Singh K, MacGregor SA (2019) Understanding electronic effects on carboxylate-assisted C-H activation at ruthenium: the importance of kinetic and thermodynamic control. Faraday Discuss 220:386–403. https://doi.org/10.1039/c9fd00063a
Davies DL, Macgregor SA, McMullin CL (2017) Computational studies of carboxylate-assisted C-H activation and functionalization at group 8–10 transition metal centers. Chem Rev 117(13):8649–8709. https://doi.org/10.1021/acs.chemrev.6b00839
Wu J, Hoang KLM, Leow ML, Liu XW (2015) Pd-catalyzed cross-coupling of aromatic compounds with carboxylic acids via C-H bond activation. Org Chem Front 2(5):502–505. https://doi.org/10.1039/C5QO00003C
Costa D, Forezi L, Lessa M, Delarmelina M, Matuck B, Freitas M, Ferreira V, Resende J, Carneiro J, Silva F (2022) A stereoselective, base‐free, palladium‐catalyzed Heck coupling between 3‐halo‐1,4‐naphthoquinones and vinyl‐1H‐1,2,3‐triazoles. ChemistrySelect 7. https://doi.org/10.1002/slct.202201334
Gaussian 09, Revision D.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima,Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M.Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J.Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R.Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian, Inc., Wallingford CT (2013)
Yanai T, Tew DP, Handy NC (2004) A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem Phys Lett 393(1–3):51–57. https://doi.org/10.1016/J.CPLETT.2004.06.011
Weigend F, Ahlrichs R (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys Chem Chem Phys 7(18):3297–3305. https://doi.org/10.1039/b508541a
Marenich AV, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113(18):6378–6396. https://doi.org/10.1021/JP810292N
Foresman JB, Frsch A (1996) Exploring chemistry with electronic structure methods, Second edition. Wallingford.
Wilson ND, Wang Z, Gung BW (2022) How to control the acidity of 1,2,3-triazolium ions: a density functional theory study. J Mol Graph Model 112. https://doi.org/10.1016/j.jmgm.2022.108133
Joseph MC, Swarts AJ, Mapolie SF (2023) Transition metal complexes of click-derived 1,2,3-triazoles as catalysts in various transformations: an overview and recent developments. Coord Chem Rev 493. https://doi.org/10.1016/j.ccr.2023.215317
Gandeepan P, Müller T, Zell D, Cera G, Warratz S, Ackermann L (2019) 3d transition metals for C-H activation. Chem Rev 119(4):2192–2452. https://doi.org/10.1021/acs.chemrev.8b00507
Pan S, Sarkar S, Ghosh B, Samanta R (2021) Transition metal catalysed direct construction of 2-pyridone scaffolds through C-H bond functionalizations. Org Biomol Chem 19(48):10516–10529. https://doi.org/10.1039/D1OB01856F
Landaeta VR, Rodríguez-Lugo RE (2017) Insights on the C-H Bond activation by transition metal complexes from groups 8–10 bearing (P-N) chelates. J Mol Catal A: Chem 426:316–325. https://doi.org/10.1016/j.molcata.2016.08.011
Walsh AP, Jones WD (2015) Mechanistic insights of a concerted metalation-deprotonation reaction with [Cp∗RhCl2]2. Organometallics 34(13):3400–3407. https://doi.org/10.1021/acs.organomet.5b00369
Wang L, Carrow BP (2019) Oligothiophene synthesis by a general C-H activation mechanism: electrophilic concerted metalation-deprotonation (eCMD). ACS Catal 9(8):6821–6836. https://doi.org/10.1021/acscatal.9b01195
Jiang H, Sun TY (2021) The activating effect of strong acid for Pd-catalyzed directed C-H activation by concerted metalation-deprotonation mechanism. Molecules 26(13):4083. https://doi.org/10.3390/molecules26134083
Slocum DW, Sugarman DI (1974) Directed metalation. 222–247. https://doi.org/10.1021/ba-1974-0130.ch012
Bouley BS, Tang F, Bae DY, Mirica LM (2023) C-H bond activation via concerted metalation–deprotonation at a palladium(III) center. Chem Sci. https://doi.org/10.1039/d3sc00034f
Funding
The authors would like to acknowledge CNPq (309080/2015–0 and 434955/2018–3), FAPERJ (E-26/203.001/2017, E-26/010.101118/2018, and E-26010.001424/2019), and the CAPES PRINT Program (88881.310460/2018–01) for financial support. Milena D. Lessa was supported by a research fellowship from the MIDAS INCT.
Author information
Authors and Affiliations
Contributions
Wagner F. Fogos: analyzed, did the bibliographic survey, and wrote the text of the article. Milena D. Lessa: critically reviewed and assisted in the writing of the text as well as in the analysis of the results obtained. José Walkimar de M. Carneiro: acted in the critical correction and direction of the work in all spheres of research and approved the version to be published. Fernando de Carvalho da Silva: worked on the experimental part to compare the data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Fogos, W.F., Lessa, M.D., de Carvalho da Silva, F. et al. Mechanistic insights into C(sp2)-H activation in 1-Phenyl-4-vinyl-1H-1,2,3-triazole derivatives: a theoretical study with palladium acetate catalyst. J Mol Model 30, 183 (2024). https://doi.org/10.1007/s00894-024-05987-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-024-05987-0