
Abstract
The strategy of investigating the antioxidant potential of flavonols through the explicit modeling of chemical reactions (initiated to be employed in a previous work from our group) was taken further in this work. Therefore, a theoretical investigation on the reaction between fisetin and 2,2-diphenyl-1-picrylhydrazyl (DPPH) is presented. All the computations were performed using the density functional theory with the B3LYP functional along with the 6-31G(d,p) basis set. Structural, energetic quantities (ΔG and ΔG++), and reaction rates were probed in order to provide information on the antioxidant activity and to explore the contributions of each hydroxyl group to the referred property. According to the results obtained for the thermodynamic properties, fisetin presents antioxidant potential similar to quercetin (behavior that is also observed experimentally). In addition, the order of contribution of each OH group to the antioxidant potential was found to be 4′-ArOH (the most contributor, presenting ΔG = -5.17 kcal/mol) → 3′-ArOH (ΔG = -3.35 kcal/mol) → 3-ArOH (ΔG = -1.64 kcal/mol) → 7-ArOH (ΔG = 7.72 kcal/mol). These observations are in consistent agreement with the outcomes of other computational investigations performed using bond dissociation enthalpies (BDEs) as descriptors for the antioxidant activity. Therefore, the methodology employed in this work can be used as an alternative for probing antioxidant potential of compounds derived from fisetin.

Illustrative scheme of the PES mapping in terms of hydrogen atom transfer from fisetin 3-ArOH to the nitrogen centered DPPH
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Flavonols are a family of organic compounds found in several families of plants. These chemical species are known for being excellent antioxidants [1,2,3] as well as for presenting anti-inflammatory and anti-cancer actions [4]. Hence, there is an ongoing focus on finding new sources of flavonols and probing the antioxidant potential of newly synthesized/isolated molecules [5, 6]. From a theoretical point of view, it is well established that the antioxidant potential of a given flavonol can be probed by the investigation of properties related with (mainly) two mechanisms: the single electron transfer (SET) and the hydrogen atom transfer (HAT) [7, 8]. The SET mechanism can be expressed as:
where R. is the free radical. In SET, the ionization potential (IP) of the molecule is important: the smaller the IP for the neutral molecule of a given flavonol (ArOH), the lower the energetic cost to abstract an electron. However, different computational investigations on several flavonols pointed the HAT mechanism as being energetically preferred over SET [9,10,11,12]. In HAT, a single step occurs such as:
where the bond dissociation enthalpies (BDEs) of the OH bonds are the most important properties. In HAT, the O-H bond from a given hydroxyl group of the flavonol is broken in a homolytic way and the respective hydrogen atom is directly transferred to the free radical. Therefore, the lower the energy to break the O-H bond, the easier it will be for the H atom to be scavenged by the free radical.
Although intrinsically thermodynamic quantities, it has been shown in a number of studies that the computationally determined BDEs are strongly correlated with experimentally measured antioxidant activity, being a good descriptor for probing the antioxidant potential of polyphenols [13]. In this scope, several studies on probing the potential antioxidant activity of flavonols through the determination of their OH BDEs have been performed in the recent past [14,15,16,17]. However, such investigations based purely on the values of BDEs do not take into account the role played by the free radical. With this in mind, very recently, our group employed a different theoretical approach to evaluate the antioxidant potential for flavonols [18]. In the referred work [18], the reactions between two well-known antioxidant compounds (quercetin and morin) and 2,2-diphenyl-1-picrylhydrazyl (DPPH), which is a free radical model widely used in experimental tests [19,20,21], were examined through a computational investigation using density functional theory (DFT). In the present work, we took the ideas initiated in Maciel et al. [18] further and performed a theoretical study on the reaction between fisetin and DPPH (both chemical structures can be seen in Fig. 1). The outcomes obtained here were discussed and compared with previous investigations that utilized BDEs for probing the HAT mechanism for fisetin. This comparison indicated that the methodology used is able to provide results that are in consistent agreement with the conclusions achieved by Vagánek et al. [22] (at the B3LYP/6-311++G∗∗ level of theory in gas phase), Marković et al. [23] (at the B3LYP/6-311++G(d,p) level of theory in gas phase), and Amić et al. [24] (using PM6 in both gas phase and water) that have considered the BDEs for investigating the antioxidant activity of fisetin. In addition, the approach utilized in the present work can give additional information about the kinetics of the hydrogen transfer. Hence, it can serve as an alternative method for probing the antioxidant potential of compounds derived from fisetin. As far as we know, this work represents the first attempt on studying the antioxidant activity of this compound considering the free radical explicitly in the calculations.

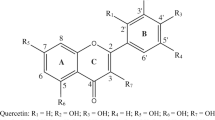
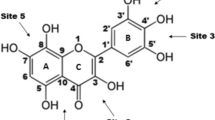
Illustration of the a fisetin and b 2,2-diphenyl-1-picrylhydrazyl (DPPH) chemical structures. Atom numbering is also presented
Computational methods
In the present work, we performed the modeling of the chemical reactions through the use of DFT with the B3LYP functional [25,26,27,28] and the 6-31G(d,p) basis set [29, 30]. The geometry of fisetin was fully optimized at the B3LYP/6-31G(d,p) level of theory. On the other hand, the unrestricted formalism (UB3LYP) combined with the same 6-31G(d,p) basis set was employed for obtaining the DPPH structure. The initial guess for the geometries of both compounds was based on previous studies [22, 31]. In order to calculate the Gibbs free-energy corrections and confirm the conformations as minima, harmonic vibrational frequencies were evaluated for each optimized structure. One by one, the reactions involving the hydrogen atoms from each OH group and the nitrogen-centered DPPH were studied considering a mechanism based on a single step (that is the case for the HAT, see Eq. 2). Hence, the optimized structures of fisetin and DPPH are put together and the potential energy surface (PES) regarding the distance between the H atom of a given hydroxyl group and the N atom of the DPPH molecule is scanned. The mapped path of the PES presented the occurrence of two minima (regarding the conformation of the reactant-complex, RC, and the possible conformation for the product-complex, PC, respectively) and a maxima (a possible transition state, TS) in between. For illustrative purposes, a schematic drawn is available in Fig. 2.

Illustrative scheme of the PES mapping in terms of hydrogen atom transfer from an hydroxyl group (3-ArOH) of fisetin to the nitrogen centered radical DPPH
The resulting PC for each reaction was optimized at the B3LYP/6-31G(d,p) level of theory (the same approach used previously for the cases of RC). In addition, quadratic synchronous transit method (QST3) was used to find the first-order saddle point and, thus, determine the TS from the structure collected at the maxima in the PES. For each reaction, the respective TS was found to have a single imaginary frequency. As several experiments for antioxidant activity with DPPH are accomplished in methanol [32,33,34], we considered this solvent environment (𝜖 = 32.613) in all the calculations performed in this work by using the integral equation formalism polarizable continuum model (IEF-PCM) [35, 36]. The Gaussian 09 suite of software [37] was utilized in all the computations performed in this work.
In order to provide some ground for comparison, previous computational results of BDEs and IPs determined by Vagánek et al. [22] (computed at the B3LYP/6-311++G∗∗ level of theory), Marković et al. [23] (determined at the B3LYP/6-311++G(d,p) level of theory), and Amić et al. [24] (obtained using the PM6 approach in both gas phase and water) were utilized.
Results and discussion
Through the analysis of the results shown in Table 1, it is possible to notice that the BDEs are considerably lower than the IP and, thus, the HAT mechanism is prevalent over SET for the antioxidant activity of fisetin, as mentioned previously. All the computational studies indicate 4′-ArOH as having the lowest BDE (in gas phase, 71.9 kcal/mol according to Vagánek et al. [22], 72.4 kcal/mol according to Marković et al. [23], and 70.3 kcal/mol according to Amić et al. [24]) followed by 3′-ArOH (74.5 kcal/mol according to Vagánek et al. [22] and 75.0 kcal/mol according to Marković et al. [23]), 3-ArOH (82.4 kcal/mol according to Vagánek et al. [22], 82.6 kcal/mol according to Marković et al. [23]), and 7-ArOH (84.6 kcal/mol according to Vagánek et al. [22], 85.0 kcal/mol according to Marković et al. [23]). This observation suggests that 4′-ArOH would be the primarily responsible for the antioxidant activity of fisetin while 7-ArOH would play a minor role in such property.
The RC for the reactions between 3′-ArOH, 4′-ArOH, 3-ArOH, and 7-ArOH of fisetin and DDPH can be seen in Fig. 3a, b, c and d, respectively. On the other hand, Fig. 4a, b, c, and d present the PC for the reactions involving DPPH and 3′-ArOH, 4′-ArOH, 3-ArOH, and 7-ArOH of fisetin, respectively. In all the cases, the coordinate of reaction investigated was the one involving the transfer of the hydrogen of a given OH group to the nitrogen radical (Nr) of DPPH. A single imaginary frequency was found for each one of the TS achieved for the four reactions studied (see all the TS structures in Fig. 5). In regard to the RC for the reaction involving 3′-ArOH and DPPH, Fig. 3a, it is possible to see the existence of a relatively strong interaction (2.07 Å) between the hydrogen atom of the 3′-ArOH and the Nr atom. Also regarding this RC, the hydrogen atom of the 4′-ArOH (neighboring the 3′-ArOH) shows an even stronger interaction (1.98 Å) with the O atom from a NO2 moiety O(nitro), which prevents the referred oxygen atom to interact with the H atom from 3′-ArOH and establishes a hydrogen interaction that is responsible for the stabilization of the RC. A similar behavior is observed in the case of the RC for the reaction between 4′-ArOH and DPPH (Fig. 3b), however with more pronounced interactions (1.99 Å between the H atom of the 4′-ArOH group and the Nr and 1.92 Å between the H atom of its neighbor (3′-ArOH) and the O(nitro) atom). As seen previously for the cases of quercetin and morin [18], these interactions provide a configuration that makes the scavenging of the H atom much more easy to occur. On the other hand, the H atom to be abstracted from 3-ArOH is presenting an interaction (about 2.17 Å) with the Nr atom from DPPH and also experiencing another interaction (about 2.13 Å) with the O(nitro) that is competing for having the H atom. The RC for the abstraction of the hydrogen atom from 7-ArOH is stabilized only by a single interaction (about 1.92 Å) between the H atom of the hydroxyl group and the Nr atom. Hence, the RC configurations for both 3-ArOH and 7-ArOH make scavenge process much more difficult to happen when in comparison to the RC of the 3′-ArOH and 4′-ArOH. In terms of the PC configurations, the reactions of the 3′-ArOH (Fig. 4a) and 4′-ArOH (Fig. 4b) with DPPH present the same four-atom interaction reported in the previous investigation for quercetin and morin [18], which contribute to the stabilization of the PC. This interaction is not found in the cases of the PC for the reactions of the 3-ArOH (Fig. 4c) and 7-ArOH (Fig. 4d) and, thus, both PC will be less stable than the PC of 3′-ArOH and 4′-ArOH.

RC configuration for fisetin-DPPH: a \({3^{\prime }}\)-ArOH, b \({4^{\prime }}\)-ArOH, c 3-ArOH, and d 7-ArOH

PC configuration for fisetin-DPPH: a \({3^{\prime }}\)-ArOH, b \({4^{\prime }}\)-ArOH, c 3-ArOH, and d 7-ArOH

TS for the reaction between: a \({3^{\prime }}\)-ArOH from fisetin and DPPH, b \({4^{\prime }}\)-ArOH from fisetin and DPPH, c 3-OH from fisetin and DPPH, and d 7-OH from fisetin and DPPH
Table 2 shows the results of Gibbs free energy (ΔG) and Gibbs free energy of activation (ΔG++) for fisetin as computed at the B3LYP/6-31G(d,p) level of theory, in methanol. ΔG and ΔG++ results for quercetin and morin taken from Maciel et al. [18] were also included in Table 2 for comparison purposes. In regards to the conformer used in the present work for fisetin, 4′-ArOH exhibited the most negative value of ΔG (-5.17 kcal/mol) among the four hydroxyl groups, suggesting that this particular OH groups would be the main contributor to the antioxidant activity. In addition, 3′-ArOH was found to have the second most negative value (ΔG = -3.35), followed by 3-ArOH (ΔG = -1.64 kcal/mol). ΔG for 7-ArOH was determined to be positive (7.72 kcal/mol) and, thus, the abstraction of the hydrogen from the referred hydroxyl will be non-spontaneous. The order of contribution to the antioxidant activity of fisetin determined with the ΔG values (4′-ArOH > 3′-ArOH > 3-ArOH > 7-ArOH) is the same as observed by Vagánek et al. [22], Marković et al. [23], and Amić et al. [24] using the approach based on BDEs. This order can also be explored in terms of highest occupied molecular orbital (HOMO) plots for all the TS, which can be found in Fig. 6. It is possible to notice, for instance, that 4′-ArOH HOMO, Fig. 6b, lies (in major part) on the DPPH molecule when compared to the 7-ArOH HOMO, Fig. 6d, in which a great part of the electronic density lies above the fisetin molecule; the lowest unoccupied molecular orbital (LUMO) is centered around the DPPH molecule in all the cases. In addition, the results of ΔG indicate fisetin being as high antioxidant as quercetin and morin, which is in agreement with experimental [38] and previous theoretical investigations [18, 22, 24]. In regards to ΔG++ for the hydroxyl groups that have shown to be thermodynamically favored (presenting ΔG < 0) in the reactions probed, it is possible to notice that 3′-ArOH presents the smallest one (ΔG++ = 7.23 kcal/mol), followed by 4′-ArOH (having ΔG++ = 9.33 kcal/mol), and 3-ArOH (with ΔG++ = 10.17 kcal/mol).

HOMO and LUMO plots for the TS of the reaction involving: a \({3^{\prime }}\)-ArOH from fisetin and DPPH, b \({4^{\prime }}\)-ArOH from fisetin and DPPH, c 3-OH from fisetin and DPPH, and d 7-OH from fisetin and DPPH
The reaction rates (determined through the use of the Eyring’s equation [39] combined with the ΔG++ values computed for all the reactions studied in this work) are presented in Table 3. The rate of the reaction between the 3′-ArOH and DPPH is approximately 35 times greater than that regarding 4′-ArOH, given that k1 = 3.14x107 and k1 = 9.01x105 for the reactions of 3′-ArOH and 4′-ArOH, respectively. On the other side, 3-ArOH presents a reaction rate 143 times smaller than 3′-ArOH. These observations indicate that the hydrogen of the 3′-ArOH will be the fastest one to be scavenged by DPPH followed by the hydrogen of the 4′-ArOH, and the hydrogen of the 3-ArOH.
Conclusions
In summary, we investigated the reaction between fisetin (a well-known antioxidant) and DPPH (a free radical widely used in antioxidant activity tests) through a computational investigation. All of the reactions were modeled by using DFT with B3LYP functional and 6-31G(d,p) basis set. Structural, energetic properties (ΔG and ΔG++), and reaction rates were probed to provide information on the antioxidant activity and to explore the contributions of each hydroxyl group. In regards to the kinetics parameters, the reaction rates are presented (from fastest to the slowest) in the following order: 3′-ArOH → 4′-ArOH → 7-ArOH → 3-ArOH. On the other hand, the thermodynamic properties indicated fisetin as being a similar antioxidant to quercetin and the order of contribution to the antioxidant potential of each OH group was determined to be 4′-ArOH (the most contributor) → 3′-ArOH → 3-ArOH → 7-ArOH. These observations are in consistent agreement with the outcomes of investigations performed using BDEs. Therefore, the methodology employed in this work can be used as an alternative for probing antioxidant potential of compounds derived from fisetin.
References
Yao Y, Lim G, Xie Y, Ma P, Li G, Meng Q, Wu T (2014) Preformulation studies of myricetin: a natural antioxidant flavonoid. Pharmazie 69:19–26
Gordon MH, Roedig-Penman A (1998) Antioxidant activity of quercetin and myricetin in liposomes. Chem Phys Lipids 97:79–85
Chobot V, Hadacek F (2011) Exploration of pro-oxidant and antioxidant activities of the flavonoid myricetin. Redox Rep 16:242–247
Nasri I, Chawech R, Girardi C, Mas E, Ferrand A, Vergnolle N, Fabre N, Mezghani-Jarraya R, Racaud-Sultan C (2017) Anti-inflammatory and anticancer effects of flavonol glycosides from Diplotaxis Harra through GSK3 beta regulation in intestinal cells. Pharm Biol 55:124–131
Bell L, Oruna-Concha MJ, Wagstaff C (2015) Identification and quantification of glucosinolate and flavonol compounds in rocket salad (Eruca sativa, Eruca vesicaria and Diplotaxis tenuifolia) by LC-MS: highlighting the potential for improving nutritional value of rocket crops. Food Chem 172:852–861
Grzesik M, Bartosz G, Dziedzic A, Narog D, Namiesnik J, Sadowska-Bartosz I (2018) Antioxidant properties of ferrous flavanol mixtures. Food Chem 268:567–576
Wright JS, Johnson ER, DiLabio GA (2001) Predicting the activity of phenolic antioxidants: theoretical method, analysis of substituent effects, and application to major families of antioxidants. J Am Chem Soc 123:1173–1183
Leopoldini M., Pitarch IP, Russo N, Toscano M (2004) Structure, conformation, and electronic properties of apigenin, luteolin, and taxifolin antioxidants. A first principle theoretical study. J Phys Chem A 108:92–96
Justino GC, Vieira AJSC (2010) Antioxidant mechanisms of quercetin and myricetin in the gas phase and in solution—a comparison and validation of semi-empirical methods. J Mol Model 16:863–876
Mohajeri A, Asemani SS (2009) Theoretical investigation on antioxidant activity of vitamins and phenolic acids for designing a novel antioxidant. J Mol Struct 930:15–20
Sadasivam K, Kumaresan R (2011) Antioxidant behavior of mearnsetin and myricetin flavonoid compounds—a DFT study. Spectrochim Acta A 79:282–293
Nenadis N, Sigalas MP (2008) A DFT study on the radical scavenging activity of maritimetin and related aurones. J Phys Chem A 112:12196–12202
Giacomelli C, Miranda FdaS, Goncalves NS, Spinelli A (2004) Antioxidant activity of phenolic and related compounds: A density functional theory study on the O-H bond dissociation enthalpy. Redox Rep 9:263–269
de Souza GLC, de Oliveira LMF, Vicari RG, Brown A (2016) A DFT investigation on the structural and antioxidant properties of new isolated interglycosidic O-(1→3) linkage flavonols. J Mol Model 22:100–109
Guajardo-Flores D, Serna-Saldivar SO, Gutiérrez-Uribe JA (2013) Evaluation of the antioxidant and antiproliferative activities of extracted saponins and flavonols from germinated black beans (Phaseolus vulgaris L.) Food Chem 141:1497–1503
Mendes RA, e Silva BLS, Takeara R, Freitas RG, Brown A, de Souza GLC (2018) Probing the antioxidant potential of phloretin and phlorizin through a computational investigation. J Mol Model 24:101
Mendes RA, Almeida SKC, Soares IN, Barboza CA, Freitas RG, Brown A, de Souza GLC (2018) A computational investigation on the antioxidant potential of myricetin 3,4′-di-O-α-L-rhamnopyranoside. J Mol Model 24:133
Maciel EN, Almeida SKC, da Silva SC, de Souza GLC (2018) Examining the reaction between antioxidant compounds and 2,2-diphenyl-1-picrylhydrazyl (DPPH) through a computational investigation. J Mol Model 24:218
Trouillas P, Marsal P, Svobovova A, Vostalova J, Gazak R, Hbrac J, Sedmera P, Kren V, Lazzaroni R, Duroux J -L, Walterova D (2008) Mechanism of the antioxidant action of silybin and 2,3-dehydrosilybin flavonolignans: A joint experimental and theoretical study. J Phys Chem A 112:1054–1063
Fezai R, Mezni A, Rzaigui M (2018) Synthesis, structural analysis, Hirshfeld surface, spectroscopic characterization and, in vitro, antioxidant activity of a novel organic cyclohexaphosphate. J Mol Struct 1154:64–71
Yang W, Fortunati E, Bertoglio F, Owczarek JS, Bruni G, Kozanecki M, Kenny JM, Torre L, Visai L, Puglia D (2018) Polyvinyl alcohol/chitosan hydrogels with enhanced antioxidant and antibacterial properties induced by lignin nanoparticles. Carbohydr Polym 181:275–284
Vagánek A, Rimarčik J, Lukeš V, Klein E (2012) On the energetics of homolytic and heterolytic O–H bond cleavage in flavonols. Comput Theor Chem 991:192–200
Marković ZS, Mentus SV, Dimitrić Marković JM (2009) Electrochemical and density functional theory study on the reactivity of fisetin and its radicals: Implications on in vitro antioxidant activity. J Phys Chem A 113:14170–14179
Amić D, Stepanić W, Lučić R, Marković Z, Dmitrić Marković J M (2013) PM6 study of free radical scavenging mechanisms of flavonoids: Why does OH bond dissociation enthalpy effectively represent free radical scavenging activity. J Mol Model 19:2593–2603
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Vosko SH, Wilk L, Nusair M (1980) Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can J Phys 58:1200–1211
Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ (1994) Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J Phys Chem 98:11623–11627
Rassolov V, Pople JA, Ratner M, Redfern PC, Curtiss LA (2001) 6-31G* basis set for third-row atoms. J Comp Chem 22:976–984
Binkley JS, Pople JA, Hehre WJ (1980) Self-consistent molecular orbital methods. 21. Small split-valence basis sets for first-row elements. J Am Chem Soc 102:939–946
Rajaraman D, Sundararajan G, Rajkumar R, Bharanidharan S, Krishnasamy K (2016) Synthesis, crystal structure investigation, DFT studies and DPPH radical scavenging activity of 1-(furan-2-ylmethyl)-2,4,5-triphenyl-1H-imidazole derivatives. J Mol Struct 1108:698–707
Miliauskas G, Venskutonis PR, van Beek TA (2004) Screening of radical scavenging activity of some medicinal and aromatic plant extracts. Food Chem 85:231–237
Kumaran A, Karunakaran RJ (2007) In vitro antioxidant activities of methanol extracts of five Phyllanthus species from India. LWT - Food Sci Technol 40:344–352
Mahdi-Pour B, Jothy SL, Latha LY, Chen Y, Sasidharan S (2012) Antioxidant activity of methanol extracts of different parts of Lantana camara. Asian Pac J Trop Biomed 2:960–965
Scalmani G, Frisch MJ (2010) Continuous surface charge polarizable continuum models of solvation. I. General formalism. J Chem Phys 132:114110
Cancès E, Mennucci B, Tomasi J (1997) A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J Chem Phys 107:3032–3041
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr. J A, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian, Inc., Wallingford CT, Gaussian 09, Revision D.01
Khan NK, Syed DN, Ahmad N, Mukhtar H (2013) Fisetin: a dietary antioxidant for health promotion. Antioxid Redox Signal 19:151–162
Atkins PW, De Paula J Físico−Química, Vol. 2, 9a.Ed., Rio de Janeiro, Brazil, LTC
Acknowledgements
This work was funded by the Brazilian agency CNPq (Process number: 306266/2016-4).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This paper belongs to the Topical Collection VII Symposium on Electronic Structure and Molecular Dynamics – VII SeedMol
Rights and permissions
About this article

Cite this article
Maciel, E.N., Soares, I.N., da Silva, S.C. et al. A computational study on the reaction between fisetin and 2,2-diphenyl-1-picrylhydrazyl (DPPH). J Mol Model 25, 103 (2019). https://doi.org/10.1007/s00894-019-3969-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-019-3969-8