
Abstract
Signaling systems allow microorganisms to sense and respond to different stimuli through the modification of gene expression. The phosphorelay signal transduction system in eukaryotes involves three proteins: a sensor protein, an intermediate protein and a response regulator, and requires the transfer of a phosphate group between two histidine-aspartic residues. The SLN1-YPD1-SSK1 system enables yeast to adapt to hyperosmotic stress through the activation of the HOG1-MAPK pathway. The genetic sequences available from Saccharomyces cerevisiae were used to identify orthologous sequences in Candida glabrata, and putative genes were identified and characterized by in silico assays. An interactome analysis was carried out with the complete genome of C. glabrata and the putative proteins of the phosphorelay signal transduction system. Next, we modeled the complex formed between the sensor protein CgSln1p and the intermediate CgYpd1p. Finally, phosphate transfer was examined by a molecular dynamic assay. Our in silico analysis showed that the putative proteins of the C. glabrata phosphorelay signal transduction system present the functional domains of histidine kinase, a downstream response regulator protein, and an intermediate histidine phosphotransfer protein. All the sequences are phylogenetically more related to S. cerevisiae than to C. albicans. The interactome suggests that the C. glabrata phosphorelay signal transduction system interacts with different proteins that regulate cell wall biosynthesis and responds to oxidative and osmotic stress the same way as similar systems in S. cerevisiae and C. albicans. Molecular dynamics simulations showed complex formation between the response regulator domain of histidine kinase CgSln1 and intermediate protein CgYpd1 in the presence of a phosphate group and interactions between the aspartic residue and the histidine residue. Overall, our research showed that C. glabrata harbors a functional SLN1-YPD1-SSK1 phosphorelay system.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The adaptation of microorganisms to different environmental conditions is regulated by different signaling pathways [1,2,3,4,5]. The response to environmental change is important for microorganism survival and involves different biological processes such as osmosensing, oxidant adaptation, light perception, morphogenesis and dimorphism, conidiation, cell wall integrity, melanin production, sporulation, expression of pathogenicity factors, and virulence [1, 6,7,8,9,10,11].
The phosphorelay signal transduction system in eukaryotes has three different proteins: a sensor protein histidine kinase (HK), an intermediate histidine phosphotransfer (HPt) protein, and a downstream response regulator (RR) protein [10, 12,13,14]. The three proteins interact through the transfer of a phosphate group from a histidine to an aspartic acid residue [10, 15, 16]. The sensor proteins in eukaryotes are hybrid, meaning that they have both HK and RR domains, and are capable of detecting environmental changes that generate the transfer of a phosphate group from ATP, which initiates the phosphorelay signal transduction system [8, 17, 18]. The HK sensor proteins are classified into 16 families, the protein Sln1 belongs to Family III characterized for their localization in the plasmatic membrane, while the intermediate HPt protein and RR are localized in the cytoplasm [18,19,20].
The phosphorelay signal transduction system regulates various processes including dimorphism, sporulation, and the expression of virulence factors, and has been studied in several human pathogenic fungi, such as, Histoplasma capsulatum, Coccidioides immitis, Paracoccidioides brasiliensis, Blastomyces dermatitidis, Sporothrix schenckii, Penicillium marneffei, Aspergillus fumigatus, Cryptococcus neoformans, Candida guilliermondii, and Candida albicans [11, 20,21,22,23].
There are intermediate proteins in fungi that harbor phosphotransferase domains; these interact with different branches of the phosphorelay signal transduction system, permitting transfer of the phosphate group between the sensor proteins and RR domains [12, 15, 24,25,26].
The phosphorelay signal transduction system in Saccharomyces cerevisiae has been studied extensively, and the interaction between the HK and RR domains of Sln1p and Ypd1p is well understood [12, 15]. It is also known that the SLN1-YPD1-SSK1 pathway regulates the response to osmotic stress while SLN1-YPD1-SKN7 regulates the response to oxidative stress [17, 27, 28].
In our research, we are interested in how the phosphorelay system works in the opportunistic yeast Candida glabrata. This yeast is phylogenetically more related to S. cerevisiae than to C. albicans [29,30,31,32]. In contrast to S. cerevisiae, C. glabrata harbors a unique putative orthologous HK gene, which encodes an Sln1p that has two functional domains: HK and RR. An in silico analysis showed that it harbors eight different transcription binding sites activated for nitrogen sources and one for Msn2/Msn4. Moreover, this gene is related to the processes of adaptation to different environmental stress conditions, such as osmotic, oxidative, pH, and the assimilation of different carbon sources [33].
The aim of this work was to look for and demonstrate in silico the functionality and interactions of the C. glabrata SLN1-YPD1-SSK1 phosphorelay system, and to understand its participation in the regulation of other signaling pathways compared with S. cerevisiae and C. albicans. We confirm the orthology of the pathway with S. cerevisiae, obtain the interactome of the different proteins, and use molecular dynamics to demonstrate phosphate transfer between the CgSln1 and CgYpd1 proteins.
Methods
Sequences
To perform the in silico analysis, the complete genome sequences of C. glabrata CBS138 held at NCBI (https://www.ncbi.nlm.nih.gov/gene/) were studied based on the reported S. cerevisiae sequences. We identified the sequence CAGL0H06567g as CgSLN1, CAGL0D02882g as CgSSK1 and CAGL0K04961g as CgYPD1. We found only one matching copy of each gene in the genome of C. glabrata. These DNA sequences were used and translated for in silico protein characterization and phylogenetic analysis. Structural modeling of the proteins was performed based on the structures 1C02 and 2R25 from the PDB (http://www.rcsb.org) of S. cerevisiae.
In silico gene and protein characterization of the C. glabrata phosphorelay signal transduction system
Once the sequences were identified, their chromosomal localization, size and coding sequences were determined. Amino acid sequences were analyzed in terms of their molecular weight and isoelectric point. The degree of identity to orthologous sequences of S. cerevisiae was determined using alignments with the PRALINE database (http://www.ibi.vu.nl/programs/pralinewww/). The activity domains of each protein were identified using the database Expasy Prosite (http://prosite.expasy.org/) [34].
Phylogenetic analysis of C. glabrata proteins Ypd1p and Ssk1p
The CgYpd1p and CgSsk1p sequences were used for phylogenetic analysis. The phylogenetic analysis of CgSln1p was reported previously [33]. Phylogeny was carried out using the Maximum Likelihood method based on the Whelan and Goldman model [35] for CgYpd1p, and the Jones-Taylor-Thornton (JTT) matrix-based model for CgSsk1p. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained by applying the neighbor-joining (NJ) method to a matrix of pairwise distances estimated using a JTT model. A discrete Gamma distribution was used to model evolutionary rate differences among sites [five categories (+G, parameter = 1.7133)]. The tree was drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved eight and nine reported Ypd1p and Ssk1p amino acid yeast sequences respectively. All positions containing gaps and missing data were eliminated. Evolutionary analyses were conducted in MEGA7 [36].
Interaction networks with the C. glabrata genome
To determine the interactions of the putative Sln1p, Ypd1p, and Ssk1p with other proteins of the C. glabrata genome, the platform STRING 9.1(http://string-db.org/) [37] was used. Most of the interacting proteins are putative so they were named based on their homology to S. cerevisiae. We found only one matching copy of each gene in the genome of S. cerevisiae.
Modeling by homology of the complex Sln1p(RR)-Ypd1 of C. glabrata
Modeling by homology was done to determine the tertiary structure of CgYpd1p and the complex CgSln1p (RR)-CgYpd1 of C. glabrata using the program MODELER 9.14 (https://salilab.org/modeller/9.14/release.html) [38]. Of the 20 different models obtained, the 5 with the lowest Discrete Optimized Protein Energy (DOPE) score were chosen [39] The structural global and local quality was evaluated on the Swiss-Model platform (http://swissmodel.expasy.org/workspace/) [40, 41], through the calculation of the atomic non-local environment assessment (ANOLEA) [42], QMEAN [43, 44] and DFire-Energy [45]. The Ramachandran plots were obtained using PROCHECK [46]. Finally, PyMOL 1.3 (https://www.pymol.org/) [47] was used to visualize structures and to calculate the root mean square deviation (RMSD).
Molecular dynamics simulation of the complex CgSln1p (RR)-CgYpd1p
A molecular dynamics simulation of the group interaction in the complex CgSln1pRR-CgYpd1 was performed. To simulate the group phosphate transfer between the RR of the sensor protein, CgSln1p and the intermediate CgYpd1p, the cubic system with the HPt-RR domains in the center was first defined. The system was solvated with water using the model TIP3P [48] and the charges were neutralized with sodium chloride to a final concentration of 0.5 M; to stabilize the complex model we added Mg++ according to Zhao et al. [49]. The system was equilibrated under a scheme NPT (310 K, 1 atm, controlled using the Langevin algorithm) through a minimization of 1000 gradient steepest-descent, followed by a 150 ps equilibration simulation with harmonic restrictions for proteins (102 kcal mol−1 Å−2). The production runs were done under the same conditions but without restrictions for 100 ns with a time step of 2 fs. The force field CHARMM27 [50] was used. Cut-off points of 10 Å were established for the van der Waals interactions and were evaluated using the particle-mesh Ewald (PME) method. The simulation was computed with NAMD 2.9 (http://www.ks.uiuc.edu/Research/namd/) [51] and the resulting trajectories were visualized and analyzed with VMD 1.9.2 [52]. For the phosphorylated complex, the phosphoaspartate parameters were taken directly from Damjanovic et al. [53], and then the force field derived CHARMM27 was used.
Results
In silico gene and protein characterization of the C. glabrata phosphorelay signal transduction system
The putative CgYpd1p (ID 2890498) was identified as being located in chromosome K, with a size of 486 bp; its translated sequence has a molecular weight of 18.5 kDa, a pI of 4.3 and 63% identity with the ScYpd1p. The putative CgSsk1p (ID 2887049) was located in chromosome D, with a size of 2004 bp; its translated sequence has a molecular weight of 75.2 kDa, a pI of 6.38 and 38.6% identity with ScSsk1p. Phylogenetic analysis of the CgYpd1p and CgSsk1p sequences were performed (Figs. 1, 2). The activity domains of each protein were identified in the sequences; CgSln1p, [33] and the CgYpd1p, and CgSsk1p harbor the HK, HPt and RR domains, respectively (Fig. 3).

Molecular phylogenetic analysis by maximum likelihood (ML) method of protein CgSsk1p of Candida glabrata (XP_445541.1) and other response regulator (RR) proteins described in Debaryomyces hansenii (XP_461427.2), Eremothecium gossypii (NP_984439.2), Kluyveromyces lactis (XP_454379.1), Lachancea thermotolerans (XP_002551906.1), Saccharomyces cerevisiae (EWG84285.1), Zygosaccharomyces rouxii (XP_002494852.1), and Candida albicans (XP_722233.1). Bar Number of nucleotide changes per 100 nucleotides. *Accession numbers from the NCBI GenBank. https://www.ncbi.nlm.nih.gov/protein/

Molecular phylogenetic analysis by ML method of protein CgYpd1p of C. glabrata (XP_448442.1) and other histidine phosphotransferase (HPt) proteins of D. hansenii (XP_458156.1), E. gossypii (NP_982765.1), K. lactis (XP_453920.1), L. thermotolerans (XP_002552137.1), S. cerevisiae (NP_010046.1), Z. rouxii (XP_002497857.1), C. albicans (XP_713887.1) and C. neoformans (XP_012053169.1). Bar Number of nucleotide changes per 100 nucleotides. *Accession numbers are from the NCBI GenBank. https://www.ncbi.nlm.nih.gov/protein/

Activity domains of each protein of the phosphorelay signal transduction system in a C. glabrata and b S. cerevisiae were identified by Expasy Prosite (http://prosite.expasy.org/) [34]. Each functional domain is marked and was observed to be conserved in both microorganisms. The CgSln1p is a hybrid protein that harbors two different domains: histidine kinase (HK) and RR, the CgYpd1p is a phosphotransfer protein that harbors HPt, and the CgSsk1p RR protein harbors a RR domain
Interaction networks of the C. glabrata phosphorelay signal transduction system
The principal interactions that mediated this assay are shown in the figures. The sensor protein HK CgSln1p (Fig 4a) and the intermediate protein CgYpd1p (Fig 4b) showed an interaction with mitochondrial proteins CgPkp1p and CgPkp2p, and with the CgSkn7, CgSsk2p, CgPbs2p and CgHog1p proteins that participate in the processes of adaptation to osmotic and oxidative stress. Further, the RR protein CgSsk1p (Fig 4c) showed interactions with the CgSte50p and CgSte11p proteins involved in cell wall synthesis. The putative proteins included in the C. glabrata interactome are shown in Table 1.

Interactions of the C. glabrata phosphorelay signal transduction system proteins. The sensor protein HK CgSln1p (Fig 4a) and the intermediate protein CgYpd1p (Fig 4b) showed an interaction with proteins that participate in processes of adaptation to osmotic and oxidative stress, while the RR protein CgSsk1p (Fig 4c) showed an interaction with proteins involved in cell wall synthesis. The interaction force (string number) was determinate using STRING 9.1
Modeling by homology of the complex Sln1p(RR)-Ypd1 of C. glabrata
We obtained 20 models of the protein CgYpd1p, from which we selected the 5 models with the lowest DOPE values. For these five models, we calculate the QMEAN, DFIRE-Energy and RMSD values. The chosen model was the one with the lowest values of all determination. The chosen model has the following characteristics: a DOPE value of −16,750.57, a DFire-Energy of −202.7, a QMEAN score of 0.567 and a RMSD of 0.208 (Online Resource 1). The model of CgYpd1 generated with MODELER is showed in Online Resource 2. The Ramachandran plots of CgYpd1 (Online Resource 3) showed an adequate clustering of the angles φ, ψ, in the allowed zones validating a high-resolution protein structure. The tertiary model showed five α-helix chains (Fig. 5a) and harbors a histidine in position 64 (Fig. 5b).

CgYpd1p surface map. a The ribbon representation is shown with the 5 α-helices labeled. b The entire surface is shown and the position of the histidine residue that receives the phosphate group is highlighted in magenta. Surface representations were created using the molecular graphics program PyMOL [41]
Further, we obtained 20 models for the CgSln1p (RR) domain. We followed the same strategy as described above. The selected model has a DOPE value of −14,265.07, a DFire-Energy of −162.25, a QMEAN score of 0.756 and a RMSD of 0.105 (Online Resource 1). The structure of CgSln1 generated with MODELER is showed in Online Resource 4. The Ramachandran plots of CgSln1p (RR) (Online Resource 5) showed an adequate clustering of the angles φ, ψ, in the allowed zones validating a high-resolution protein structure. The aspartic residue that transfers the phosphate group to the residue histidine of CgYpd1p was located (Fig. 6a). A model of the complex CgSln1p (RR)-CgYpd1p was obtained based on both structures described above and the aspartate residue 114 and histidine 64 that transfer the phosphate group were located, and are shown in Fig 6b.

CgSln1p (RR) surface map. a The ribbon representation is shown and the aspartic residue is labeled with blue spheres. b Complex CgSln1p (RR)-CgYpd1p surface map. The ribbon representation is shown and the aspartic 114 residue and histidine 64 residue, which are involved in the phosphate group transfer are labeled with magenta spheres. Surface representations were created using the molecular graphics program PyMOL [41]
Simulation of phosphate group transfer by the complex CgSln1p (RR)-CgYpd1p using molecular dynamics simulations
Molecular dynamics between the RR domain of the sensor protein HK CgSln1p and the HPt domain of the intermediate protein CgYpd1p were obtained. The CgSln1p (RR)-CgYpd1 complex was, in general, stable (RMSD below 4 Å). The RMSD of the structure Sln1RR-Ypd1 was calculated relative to the initial structure, aligning all frames using the backbone with the VMD software. In the dynamic “without phosphate” group, we obtained an average RMSD value of 2.405, and the value of the “with phosphate” group was 3.058. We present the performance of the RMSD through frames as supplementary material (Online Resources 6 and 7 respectively). We evaluated the interaction between the aspartic–histidine complex by determining the distances by 100 ns in the presence and absence of a phosphate group (Fig. 7). In the absence of a phosphate group, we observed variable distances between histidine and aspartic residues in the range of 3 Å to 10 Å (Fig. 8a; Online Resource 8), while, in the presence of the phosphate group, these residues have a stable interaction after 10 ns with a distance of 2 Å that was stable for at least 80 ns (Fig. 8b; Online Resource 9).

Graphical representation of the interaction between the aspartic residue of the CgSln1p (RR) domain and the histidine residue of the CgYpd1p protein in (a) absence and (b) presence of the phosphate group. Representations were created using the molecular graphics program VMD [44]

Measurement of the distances between the aspartic residue of the CgSln1p (RR) domain and the histidine residue of CgYpd1p during the transfer simulation in (a) absence and (b) presence of the phosphate group
We also analyzed the interaction of other conserved amino acids between the CgYpd1p and CgSln1p RR and found that several types of interactions between both proteins such as hydrogen bridges, ionic bridges, hydrophobic interactions and molecular rearrangements exist to allow the interaction between the histidine and aspartic acid (Table 2).
Discussion
The yeast C. glabrata is commensal in healthy people; however, recently it has been considered an opportunistic pathogen in immunocompromised patients. C. albicans and C. glabrata are the two most common Candida species found in the human digestive tract, and are responsible for 65–75% of systemic candidiasis, with high morbidity and mortality [54, 55]. The microorganisms harbor signal transduction pathways that mediate adaptation to extracellular environments, and which control transcriptional programs and posttranscriptional processes that modify cell metabolism to maintain homeostasis [56]. One of the most studied signaling systems is the two-component system (TCS), which is widely distributed in bacteria, plants, and eukaryotes. In fungi, it is known as the phosphorelay signal transduction system, because its main function of transferring a phosphate group is maintained but more than two proteins are involved [1, 5, 57,58,59].
C. glabrata is more related phylogenetically to S. cerevisiae than to other Candida species such as C. albicans. Our phylogenetic analysis based on the protein sequences of the RR protein CgSsk1p (Fig. 1) and the intermediate protein CgYpd1p (Fig. 2) confirm the close relationship with S. cerevisiae and its separation with C. albicans [60, 61], as well as the orthology of such proteins. Similar results were obtained previously for the sensor protein CgSln1p [33]. Moreover, the putative proteins of C. glabrata harbor the typical domains that are conserved in the S. cerevisiae phosphorelay system (Fig. 3); these domains were identified as HK and RR for CgSln1p, a phosphotransferase domain for CgYpd1, and a RR domain for CgSsk1p, as has been reported for various yeasts in addition to S. cerevisiae, such as Schizosaccharomyces pombe, C. albicans, C. neoformans and Kluyveromyces lactis [56].
We used the CgSln1p, CgYpd1p, and CgSsk1p sequences from C. glabrata to obtain the interactomes of these proteins with others encoded in the C. glabrata genome. The interactome revealed the connection with proteins from the MAPKk pathway. The MAPK pathways are used by eukaryotes for intracellular signaling transduction. In yeast, there are five pathways: pheromone response, filamentation/invasion, high osmolarity glycerol HOG1, cell integrity, and spore wall assembly [62]. We found an interaction of CgSln1p through CgYpd1p with CgSsk1p, CgSsk2p, and CgHog1p, while CgHog1p interacts with CgSho1p (Fig. 4a), and all these proteins regulated the HOG1 pathway in S. cerevisiae. This pathway allows for adaptation to osmotic stress under normal osmolarity. CgSln1p, CgYpd1p, and CgSsk1p form a phosphorylation chain, maintaining an inactive MAPK pathway (Ssk2p, Ssk22p, and Pbs2p). However, when the cells are exposed to hyperosmolar conditions, the sensor protein CgSln1p detects the environmental change, interrupting the transfer of the phosphate group to the intermediate protein CgYpd1p, and the RR CgSsk1p. This interrupts the accumulation of dephosphorylated CgSsk1p, allowing its interaction with CgSsk2p, CgSsk22p, and CgPbs2p, which induces the activation of HOG1pathway and the overproduction of glycerol and inducing the adaptation to hyperosmolarity [63,64,65,66,67]. The presence of these proteins and the predicted interactions suggest that this pathway has the same function in C. glabrata as in S. cerevisiae. These results confirm the findings of Gregori et al. [9] that the HOG1 pathway in C. glabrata can be activated by CgSln1p and CgSho1p. It has also been reported that CgSho1p is activated by the stress induced by organic acids [9]. Further, Guzmán-González et al. [33] reported that the expression of CgSLN1 is downregulated by osmotic stress. Based on all these data, we infer that the activation of the HOG1 pathway in C. glabrata is related to osmotic stress. Interestingly, in C. albicans, HOG1 is activated by CaSln1p, which regulates the adaptation to osmotic stress and the expression of virulence factor as the white-opaque phenotypic switch that enables this pathogenic yeast to evade the host immune response [66, 68]. More research is needed to clarify whether the C. glabrata phosphorelay system is involved in its pathogenesis.
Another interesting interaction was found among the transcription factor CgSkn7p and the proteins CgSln1, CgYpd1, and CgSsk1 (Fig. 4). The S. cerevisiae Skn7 transcription factor regulates adaptation to oxidative and osmotic stresses through activation of the SLN1-YPD1-SKN7 pathway [28] and is also related to the synthesis of cell wall components [69]. Further, we observed the interaction of the proteins CgYpd1 (Fig. 4b) and CgSsk1 (Fig. 4c) with different intermediate proteins from other MAP kinases pathways such as the transcription factors CgSte11, CgSte50, and CgSte7, which participate in the filamentation growth/invasion and mating pheromone response pathways in S. cerevisiae, S. pombe, and C. neoformans [62, 70, 71]. The filamentation growth and the invasion process have been related to starvation as this response allows the relocation of yeast to zones with higher nutrient concentration [72]. The change to filamentation growth requires remodeling in the cell wall composition, and the transcription factors CgSte11, CgSte50, and CgSte7 also participate here. In contrast C. glabrata does not present filamentation growth, and the pseudohyphal formation has been reported under specific nitrogen sources, and in vitro [73]; moreover, until now a sexual cycle in C. glabrata has been not described, despite the presence of the necessary genes [74, 75]. Therefore, the transcription factors CgSte11, CgSte50, and CgSte7 could be regulating the cell wall composition in response to environmental stress.
Other proteins that interact with CgYpd1 (Fig. 4b) were CgPkp2p and CgPkp1p, which are mitochondrial kinases that regulate the activity of the pyruvate dehydrogenase complex in S. cerevisiae [76, 77]. This regulation could be very relevant for the metabolism of C. glabrata because this yeast has suffered a strong genomic reduction of mostly metabolic genes. Indeed, it is only able to metabolize glucose and trehalose, and can realize fermentation even in aerobic conditions [78].
Our studies suggest that CgSln1 can act as a sensor protein for several pathways, including the pheromone response pathway, filamentation/invasion pathway, high osmolarity glycerol pathway, cell integrity, and spore wall assembly pathway. Additionally, we detected a single intermediate CgYpd1 that is able to interact with several response regulators (CgSkn7, CgSte50, CgSte11, CgSsk1, CgSsk22, and CgSte7) allowing the adequate response to different environmental conditions (Fig. 4b), which could be one of the reasons why C. glabrata is a very successful opportunistic pathogen, similar to C. albicans and C. neoformans [79,80,81].
In order to perform this analysis, we used the information from S. cerevisiae. We selected reported genes from the phosphorelay system, and we looked for homologous sequences in C. glabrata genome. We found only single homologous sequences in C. glabrata genome, both for the main proteins involved in the phosphorelay system, as well for those detected in the interactome. Several authors suggest co-evolution during the analysis of the phosphorelay systems of bacteria [82, 83] and yeast [84]. This phenomenon could explain the detection of S. cerevisiae orthologous sequences interacting with the phosphorelay system of C. glabrata.
The intermediate response protein of C. glabrata CgYpd1 has the capacity to interact with several RR. We next analyzed the structure of the proteins in more detail. Homology modeling using the information of the crystallized Ypd1p from S. cerevisiae [85, 86] was employed to obtain a model of the CgYpd1 (Fig. 5a). We observed the presence of 4 α-helix and the histidine residue 64, which are both typically found in intermediate proteins with a phosphotransferase domain [15]. This result supports the finding that CgYpd1 is in fact the intermediate protein that participates in the C. glabrata phosphorelay signaling pathway. S. cerevisiae also contains ScYpd1p with histidine residue 64, and several other positions that stabilize the structure of the intermediate protein, which allows the interaction with the receptor protein or the RR as ScSsk1p, ScSkn7p (Fig. 5b) [12, 49]. Using this information, we constructed a molecular homology model using the complex of S. cerevisiae ScSln1 (RR)-ScYpd1 to evaluate in silico the interaction between the CgSln1 (RR) domain and CgYpd1p (Fig. 6). We evaluated the interaction between the functional aspartic 114 of the CgSln1 (RR) and the histidine 64 in CgYpd1 in the absence and presence of a phosphate group (Fig. 7). We found that the distances between the aspartic and the histidine residues had a value of 3–10 Å in the absence of the phosphate group (Fig. 8a; Online Resource 8); while the distance between the two amino acid residues was 2–4 Å in its presence (Fig. 8b; Online Resource 9). This shorter distance suggests a closer interaction due to the transference of the phosphate group. In S. cerevisiae, in the absence of stress, the sensor protein ScSln1 autophosphorylates and maintains a phosphorylation chain between the residues His-Asp through the proteins Sln1-Ypd1-Ssk1. In contrast, under a stress condition, the phosphorylation chain breaks, allowing the proteins ScYpd1 and ScSsk1 to interact with other regulator proteins, which permits adaptation through the activation of the HOG1 pathway [87,88,89]. Moreover, our analysis showed the presence of nine conserved CgYpd1 amino acids, in comparison to those from ScYpd1 that interact with the CgSln1RR domain [90]. (Table 2).
One of the most interesting things about the phosphorelay system is the understanding of how the phosphorylation rates controls environmental stimuli. Previously Janiak-Spens et al. [91] evaluated the rate of phosphoryl transfer between the RR-Sln1 and Ypd1 from S. cerevisiae; they found it relatively fast, contrasting with the speed rate of the Bacillus subtilis sporulation controller phosphorelay system [92], and slower than that of Escherichia coli, which control chemotaxis [93]. Those results suggested that transfer speed rate reflects the function of the signal transduction system. Our research demonstrates a very high homology in structure and function between the phosphorelay system of S. cerevisiae and C. glabrata; thus, we hypothesize a phosphate transfer speed rate in C. glabrata similar to that in S. cerevisiae.
Our results suggest that C. glabrata harbors a functional phosphorelay system that is related to the one in S. cerevisiae. Our interactomes suggest that this system regulates adaptation to osmotic and oxidative stress and cell wall synthesis. Our analysis also showed that this system participates in the pheromone response, mating and sexual cycle, these activities are not present in C. glabrata, although the genes are present. The next step in our research is to obtain a C. glabrata (sln1Δ) to confirm the functionality of this system in C. glabrata.
Summary
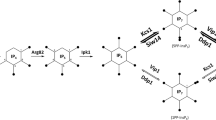
The proteins of the C. glabrata phosphorelay signaling system were identified using available databases. The intermediate protein CgYpd1p and the RR protein CgSsk1 are orthologues to the corredponding proteins in the ascomycete S. cerevisiae. Further, we identified the following functional domains: HK and RR in the sensor protein CgSln1, the HPt domain in the intermediate protein CgYpd1p and the RR domain in the protein CgSsk1. Based on the interactome obtained, the tertiary structure of the CgYpd1 protein and the interaction with the CgSln1p (RR) domain observed in the in silico dynamic molecular assay, we hypothesize that the phosphorelay signaling system is related to several signaling pathways that participate in different processes of adaptation to the environment. In Fig. 9, we highlight the findings of this in silico research; under normal conditions, the SLN1-YPD1-SSK1 system is phosphorylated and inactive, without interaction with other proteins (Fig. 9a), but when an environment change occurs, the CgSln1p senses a signal that causes dephosphorylation and uncouples the system, CgYpd1p interacts with CgSkn7p and regulates oxidative stress, CgSte11p and CgSte7 regulate cell wall composition and biosynthesis, and CgSte20 and CgSte50 regulate the mating and cell cycle, which was not detected in C. glabrata. CgSsk1p interacts with CgSsk2p, CgSsk22p, CgPbs2p, and CgHog1p to regulate the adaptation to osmotic stress. This adaptation is also regulated by CgSho1p (Fig. 9b). This adaptation capability of different pathways enables C. glabrata to be a successful opportunistic pathogen. The next step in our research is to confirm these in silico findings by obtaining mutants for CgSln1p or CgYpd1p genes to corroborate its participation in the adaptation to osmotic stress, oxidative stress, cell wall biosynthesis, and its possible role in the pathogenicity of this opportunistic yeast.

Schematic representation of the possible interaction of the C. glabrata SLN1-YPD1-SSK1 phosphorelay signaling system with other MAPKk pathways. All the proteins were detected in the interactome and named based on their homology to orthologous proteins in S. cerevisiae. a Phosphorelay system under normal conditions. b Interaction through a phosphorylation chain with other MAPKk pathways
References
Alex LA, Korch C, Selitrennikoff CP, Simon MI (1998) COS1, a two-component histidine kinase that is involved in hyphal development in the opportunistic pathogen Candida albicans. Proc Natl Acad Sci USA 95:7069–7073
Hohmann S, Krantz M, Nordlander B (2007) Yeast osmoregulation. Meth Enzymol 428:29–45
Hersen P, McClean MN, Mahadevan L, Ramanathan S (2008) Signal processing by the HOG MAP kinase pathway. Proc Natl Acad Sci USA 105:7165–7170. https://doi.org/10.1073/pnas.0710770105
Whitworth DE, Cock PJ (2009) Evolution of prokaryotic two-component systems: insights from comparative genomics. Amino Acids 37:459–466. https://doi.org/10.1007/s00726-009-0259-2
Szurmant H, Hoch JA (2010) Interaction fidelity in two-component signaling. Curr Opin Microbiol 13:190–197. https://doi.org/10.1016/j.mib.2010.01.007
Alonso-Monge R, Navarro-García F, Molero G, Diez-Orejas R, Gustin M, Pla J, Sánchez M, Nombela C (1999) Role of the mitogen-activated protein kinase Hog1p in morphogenesis and virulence of Candida albicans. J Bacteriol 181(10):3058–3068
Alonso-Monge R, Navarro-García F, Román E, Negredo AI, Eisman B, Nombela C, Pla J (2003) The Hog1 mitogen-activated protein kinase is essential in the oxidative stress response and chlamydospore formation in Candida albicans. Eukaryot Cell 2:351–361. https://doi.org/10.1128/EC.2.2.351-361.2003
Catlett NL, Yoder OC, Turgeon BG (2003) Whole-genome analysis of two-component signal transduction genes in fungal pathogens. Eukaryot Cell 2:1151–1161. https://doi.org/10.1128/EC.2.6.1151-1161.2003
Gregori C, Schüller C, Roetzer A, Schwarzmüller T, Ammerer G, Kuchler K (2007) The high-osmolarity glycerol response pathway in the human fungal pathogen Candida glabrata strain ATCC 2001 lacks a signaling branch that operates in Baker’s yeast. Eukaryot Cell 6:1635–1645. https://doi.org/10.1128/EC.00106-07
Casino P, Rubio V, Marino A (2010) The mechanism of signal transduction by two-component systems. Curr Opin Struct Biol 20:763–771. https://doi.org/10.1016/j.sbi.2010.09.010
Hérivaux A, So YS, Gastebois A, Latgé JP, Bouchara JP, Bahn YS, Papon N (2016) Major sensing proteins in pathogenic fungi: the hybrid histidine kinase family. PLoS Pathog 12(7):e1005683. https://doi.org/10.1371/journal.ppat.1005683
Xu Q, Porter SW, West AH (2003) The yeast YPD1/SLN1 complex: insights into molecular recognition in two-component signaling systems. Structure 11:1569–1581. https://doi.org/10.1016/j.str.2003.10.016
Kruppa M, Calderone R (2006) Two-component signal transduction in human fungal pathogens. FEMS Yeast Res 6:149–159. https://doi.org/10.1111/j.1567-1364.2006.00024.x
Li D, Agrellos OA, Calderone R (2010) Histidine kinases keep fungi safe and vigorous. Curr Opin Microbiol 13:424–430. https://doi.org/10.1016/j.mib.2010.04.007
Janiak SF, Cook PF, West AH (2005) Kinetic analysis of YPD1-dependent phosphotransfer reactions in the yeast osmoregulatory phosphorelay system. Biochemistry 44:377–386. https://doi.org/10.1021/bi048433s
Fassler JS, West AH (2013) Histidine phosphotransfer proteins in fungal two-component signal transduction pathways. Eukaryot Cell 12:1052–1060. https://doi.org/10.1128/EC.00083-13
Li S, Ault A, Malone CL, Raitt D, Dean S, Johnston LH, Deschenes RJ, Fassler JS (1998) The yeast histidine protein kinase, Sln1p, mediates phosphotransfer to two response regulators, Ssk1p and Skn7p. EMBO J 17:6952–6962. https://doi.org/10.1093/emboj/17.23.6952
Capra EJ, Laub MT (2012) The evolution of two-component signal transduction systems. Annu Rev Microbiol 66:325–347. https://doi.org/10.1146/annurev-micro-092611-150039
Smith DA, Morgan BA, Quinn J (2010) Stress signaling to fungal stress activated protein kinase pathways. FEMS Microbiol Lett 306(1):1–8. https://doi.org/10.1111/j.1574-6968.2010.01937
Foureau E, Clastre M, Obando ME, Besseau S, Oudin A, Glévarec G, Simkin AJ, Créche J, Atehortúa L, Giglioli-Guivarch N, Courdavault V, Papon N (2014) Subcellular localization of the histidine kinase receptors Sln1p, Nik1p and Chk1p in the yeast CTG clade species Candida guilliermondii. Fungal Genet Biol 65:25–36. https://doi.org/10.1016/j.fgb.2014.01.007
Hagiwara D, Takahashi-Nakaguchi A, Toyotome T, Yoshimi A, Abe K, Kamei K, Gonoi T, Kawamoto S (2013) NikA/TcsC histidine kinase is involved in conidiation, hyphal morphology, and responses to osmotic stress and antifungal chemicals in Aspergillus fumigatus. PLoS One 8(12):e80881. https://doi.org/10.1371/journal.pone.0080881
Valiante V, Macheleidt J, Föge M, Brakhage A (2015) The Aspergillus fumigatus cell wall integrity signaling pathway: drug target, compensatory pathways, and virulence. Front Microbiol 6:325. https://doi.org/10.3389/fmicb.2015.00325
Defosse TA, Sharma A, Mondal AK, Dugé de Bernonville T, Latgé JP, Calderone R, Giglioli-Guivarc'h N, Courdavault V, Clastre M, Papon N (2015) Hybrid histidine kinases in pathogenic fungi. Mol Microbiol 95:914–924. https://doi.org/10.1111/mmi.12911
Song HK, Lee JY, Lee MG, Moon J, Min K, Yang JK, Suh SW (1999) Insights into eukaryotic multistep phosphorelay signal transduction. J Mol Biol 293:753–761. https://doi.org/10.1006/jmbi.1999.3215
Calera JA, Herman D, Calderone R (2000) Identification of YPD1 a gene of Candida albicans which encodes a two-component phosphohistidine intermediate protein. Yeast 16:1053–1059. https://doi.org/10.1002/1097-0061(200008)16:11<1053::AID-YEA598>3.0.CO;2-H
West AH, Stock AM (2001) Histidine kinases and response regulator proteins in two-component signaling systems. Trends Biochem Sci 26:369–376. https://doi.org/10.1016/S0968-0004(01)01852-7
Brewster JL, De Valoir T, Dwyer ND, Winter E, Gustin MC (1993) An osmosensing signal transduction pathway in yeast. Science 260:1760–1762. https://doi.org/10.1126/science.7681220
Mulford KE, Fassler JS (2011) Association of the Skn7 and Yap1 transcription factors in the Saccharomyces cerevisiae oxidative stress response. Eukaryot Cell 10:761–769. https://doi.org/10.1128/EC.00328-10
Kaur R, Domergue R, Zupancic ML, Cormack BP (2005) A yeast by any other name: Candida glabrata and its interaction with the host. Curr Opin Microbiol 8:378–384. https://doi.org/10.1016/j.mib.2005.06.012
Roetzer A, Gabaldón T, Schüller C (2011) From Saccharomyces cerevisiae to Candida glabrata in a few easy steps: important adaptations for an opportunistic pathogen. FEMS Microbiol Lett 314:1–9. https://doi.org/10.1111/j.1574-6968.2010.02102.x
Jandric Z, Schüller C (2011) Stress response in Candida glabrata: pieces of a fragmented picture. Future Microbiol 6:1475–1484. https://doi.org/10.2217/fmb.11.131
Rodrigues CF, Silva S, Henriques M (2014) Candida glabrata: a review of its features and resistance. Eur J Clin Microbiol Infect Dis. 33:673–688. https://doi.org/10.1007/s10096-013-2009-3
Guzmán-González KD, Parra-Ortega B, Martínez-Rivera MA, Hernández-Rodríguez C, Pérez NO, Rodríguez-Tovar A (2013) Investigating the role of Candida glabrata SLN1 gene in stress adaptation: in silico and molecular analysis. Afr J Microbiol Res https://doi.org/10.5897/AJMR12.1448
de Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, Hulo N (2006) ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res 34:W362–W365. https://doi.org/10.1093/nar/gkl124
Whelan S, Goldman N (2001) A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol 18:691–699. https://doi.org/10.1093/oxfordjournals.molbev.a003851
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ (2013) STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 41:D808–D815. https://doi.org/10.1093/nar/gks1094
Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A (2007) Comparative protein structure modeling using Modeller. Curr Protoc Protein Sci. https://doi.org/10.1002/0471140864.ps0209s50
Eramian D, Eswar N, Shen M-Y, Sali A (2008) How well can the accuracy of comparative protein structure models be predicted? Protein Sci 17:1881–1893. https://doi.org/10.1110/ps.036061.108
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201. https://doi.org/10.1093/bioinformatics/bti770
Kiefer F, Arnold K, Künzli M, Bordoli L, Schwede T (2009) The SWISS-MODEL repository and associated resources. Nucleic Acids Res 37:D387–D392. https://doi.org/10.1093/nar/gkn750
Melo F, Devos D, Depiereux E, Feytmans E (1997) ANOLEA: a www server to assess protein structures. ISMB 5:187–190
Melo F, Feytmans E (1998) Assessing protein structures with a non-local atomic interaction energy. J Mol Biol 277:1141–1152
Benkert P, Tosatto S, Schomburg D (2008) QMEAN: a comprehensive scoring function for model quality assessment. Proteins 71:261–277. https://doi.org/10.1002/prot.21715
Zhou H, Zhou Y (2002) Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci 11:2714–2726. https://doi.org/10.1110/ps.0217002
Laskowski R, MacArthur M, Moss D, Thornton J (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26:283–291
Schrödinger, LLC (2010) The PyMOL molecular graphics system, version 1.3r1. Schrödinger, LLC, New York
Sun Y, Kollman PA (1995) Hydrophobic solvation of methane and nonbond parameters of the TIP3P water model. J Comput Chem 16:1164–1169. https://doi.org/10.1002/jcc.540160910
Zhao X, Copeland DM, Soares AS, West AH (2008) Crystal structure of a complex between the phosphorelay protein YPD1 and the response regulator domain of SLN1 bound to a phosphoryl analog. J Mol Biol 375:1141–1151. https://doi.org/10.1016/j.jmb.2007.11.045
Mackerell Jr AD, Feig M, Brooks CL (2004) Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem 25:1400–1415. https://doi.org/10.1002/jcc.20065
Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802. https://doi.org/10.1002/jcc.20289
Humphrey W, Dalke A, Schulten K (1996) VMD—visual molecular dynamics. J Mol Graph 14:33–38. https://doi.org/10.1016/0263-7855(96)00018-5
Damjanovic A, García-Moreno BE, Brooks BR (2009) Self-guided Langevin dynamics study of regulatory interactions in NtrC. Proteins 76:1007–1019. https://doi.org/10.1002/prot.22439
Kirpich IA, Solovieva NV, Leikhter SN, Shidakova NA, Lebedeva OV, Sidorov PI, Cave M (2008) Probiotics restore bowel flora and improve liver enzymes in human alcohol-induced liver injury: a pilot study. Alcohol 42:675–682. https://doi.org/10.1016/j.alcohol.2008.08.006
Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker R, Gill SR (2013) Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology 57:601–609. https://doi.org/10.1002/hep.26093
Salas-Delgado G, Ongay-Larios L, Kawasaki-Watanabe L, López-Villaseñor I, Coria R (2017) The yeasts phosphorelay systems: a comparative view. World J Microbiol Biotechnol 33:111. https://doi.org/10.1007/s11274-017-2272-z
Ota MI, Varshavsky A (1993) A yeast protein similar to bacterial two-component regulators. Science 262:566–569 http://www.jstor.org/stable/2882581
Posas F, Wurgler-Murphy SM, Maeda T, Witten EA, Thai TC, Saito H (1996) Yeast HOG1 MAP kinase cascade is regulated by a multistep phosphorelay mechanism in the SLN1-YPD1-SSK1 "two-component" osmosensor. Cell 86:865–875. https://doi.org/10.1016/S0092-8674(00)80162-2
Nemecek JC, Wüthrich M, Klein BS (2006) Global control of dimorphism and virulence in fungi. Science 312:583–588. https://doi.org/10.1126/science.1124105
Gabaldón T, Martin T, Marcet-Houben M, Durrens P, Bolotin-Fukuhara M, Lespinet O, Bouchier C (2013) Comparative genomics of emerging pathogens in the Candida glabrata clade. BMC Genomics. https://doi.org/10.1186/1471-2164-14-623
Turner SA, Butler G (2014) The Candida pathogenic species complex. Cold Spring Harb Perspect Med 4(9):a019778. https://doi.org/10.1101/cshperspect.a019778
Davenport KD, Williams KE, Ullmann BD, Gustin MC (1999) Activation of the Saccharomyces cerevisiae filamentation/invasion pathway by osmotic stress in high-osmolarity glycogen pathway mutants. Genetics 153:1091–1103
Wurgler-Murphy SM, Maeda T, Witten EA, Saito H (1997) Regulation of the Saccharomyces cerevisiae HOG1 mitogen-activated protein kinase by the PTP2 and PTP3 protein tyrosine phosphatases. Mol Cell Biol 17:1289–1297. https://doi.org/10.1128/MCB.17.3.1289
O’Rourke SM, Herskowitz I (1998) The Hog1 MAPK prevents cross talk between the HOG and pheromone response MAPK pathways in Saccharomyces cerevisiae. Genes Dev 12:2874–2886. https://doi.org/10.1101/gad.12.18.2874
Kapteyn JC, Ter Riet B, Vink E, Blad S, De Nobel H, Van Den Ende H, Klis FM (2001) Low external pH induces HOG1-dependent changes in the organization of the Saccharomyces cerevisiae cell wall. Mol Microbiol 39:469–480. https://doi.org/10.1046/j.1365-2958.2001.02242.x
Cheetham J, Smith DA, da Silva DA, Doris KS, Patterson MJ, Bruce CR, Quinn J (2007) A single MAPKKK regulates the Hog1 MAPK pathway in the pathogenic fungus Candida albicans. Mol Biol Cell 18:4603–4614. https://doi.org/10.1091/mbc.E07-06-058
Murakami Y, Tatebayashi K, Saito H (2008) Two adjacent docking sites in the yeast Hog1 mitogen-activated protein (MAP) kinase differentially interact with the Pbs2 MAP kinase kinase and the Ptp2 protein tyrosine phosphatase. Mol Cell Biol 28:2481–2494. https://doi.org/10.1128/MCB.01817-07
Liang SH, Cheng JH, Deng FS, Tsai PA, Lin CH (2014) A novel function for Hog1 stress-activated protein kinase in controlling white-opaque switching and mating in Candida albicans. Eukaryot Cell 13:1557–1566. https://doi.org/10.1128/EC.00235-14
Krishna M, Narang H (2008) The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cell Mol Life Sci 65:3525–3544. https://doi.org/10.1007/s00018-008-8170-7
Matsuyama A, Yabana N, Watanabe Y, Yamamoto M (2000) Schizosaccharomyces pombe Ste7p is required for both promotion and withholding of the entry to meiosis. Genetics 155:539–549
Erdman S, Snyder M (2001) A filamentous growth response mediated by the yeast mating pathway. Genetics 159:919–928
Wang Y, Dohlman HG (2002) Pheromone-dependent ubiquitination of the mitogen-activated protein kinase kinase Ste7. J Biol Chem 277:15766–15772. https://doi.org/10.1074/jbc.M111733200
Csank C, Haynes K (2000) Candida glabrata displays pseudohyphal growth. FEMS Microbiol Lett 189:115–120. https://doi.org/10.1111/j.1574-6968.2000.tb09216.x
Wong S, Fares MA, Zimmermann W, Butler G, Wolfe KH (2003) Evidence from comparative genomics for a complete sexual cycle in the 'asexual' pathogenic yeast Candida glabrata. Genome Biol 4(2): R10. https://doi.org/10.1186/gb-2003-4-2-r10
Butler G, Rasmussen MD, Lin MF, Santos MA, Sakthikumar S, Munro CA et al (2009) Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature 459:657–662. https://doi.org/10.1038/nature08064
Pronk JT, de Steensma YH, van Dijken JP (1996) Pyruvate metabolism in Saccharomyces cerevisiae. Yeast 12:1607–1633. https://doi.org/10.1002/(SICI)1097-0061(199612)12:16<1607::AID-YEA70>3.0.CO;2-4
Gey U, Czupalla C, Hoflack B, Rödel G, Krause-Buchholz U (2008) Yeast pyruvate dehydrogenase complex is regulated by a concerted activity of two kinases and two phosphatases. J Biol Chem 283:9759–9767. https://doi.org/10.1074/jbc.M708779200
Roy S, Thompson D (2015) Evolution of regulatory networks in Candida glabrata: learning to live with the human host. FEMS Yeast Res 15(8):pii: fov087. https://doi.org/10.1093/femsyr/fov087
Lee JW, Ko YJ, Kim SY, Bahn YS (2011) Multiple roles of Ypd1 phosphotransfer protein in viability, stress response, and virulence factor regulation in Cryptococcus neoformans. Eukaryot Cell 10:998–1002. https://doi.org/10.1128/EC.05124-11
Mavrianos J, Desai C, Chauhan N (2014) Two-component histidine phosphotransfer protein Ypd1 is not essential for viability in Candida albicans. Eukaryot Cell 13:452–460. https://doi.org/10.1128/EC.00243-13
Kennedy EN, Menon SK, West AH (2016) Extended N-terminal region of the essential phosphorelay signaling protein Ypd1 from Cryptococcus neoformans contributes to structural stability, phosphostability and binding of calcium ions. FEMS Yeast Res 16(6):pii: fow068. https://doi.org/10.1093/femsyr/fow06
Schug A, Weigt M, Onuchic JN, Hwa T, Szurmant H (2009) High-resolution protein complexes from integrating genomic information with molecular simulation. Proc Natl Acad Sci USA (52):22124–22129. doi: https://doi.org/10.1073/pnas.0912100106
Dago AE, Schug A, Procaccini A, Hoch JA, Weigt M, Szurmant H (2012) Structural basis of histidine kinase autophosphorylation deduced by integrating genomics, molecular dynamics, and mutagenesis. Proc Natl Acad Sci USA 109(26):E1733–E1742. https://doi.org/10.1073/pnas.1201301109
Cheng RR, Morcos F, Levine H, Onuchic JN (2014) Toward rationally redesigning bacterial two-component signaling systems using coevolutionary information. Proc Natl Acad Sci USA 111(5):E563–E571. https://doi.org/10.1073/pnas.1323734111
Xu Q, West AH (1999) Conservation of structure and function among histidine-containing phosphotransfer (HPt) domains as revealed by the crystal structure of YPD1. J Mol Biol 292:1039–1050. https://doi.org/10.1006/jmbi.1999.3143
Waldburger CD (2003) His-asp signal transduction via a monomeric histidine phosphotransfer protein. Structure 11:1461–1462. https://doi.org/10.1016/j.str.2003.11.014
Janiak SF, West AH (2000) Functional roles of conserved amino acid residues surrounding the phosphorylatable histidine of the yeast phosphorelay protein YPD1. Mol Microbiol 37:136–144. https://doi.org/10.1046/j.1365-2958.2000.01973.x
Janiak SF, Sparling DP, West AH (2000) Novel role for an HPt domain in stabilizing the phosphorylated state of a response regulator domain. J Bacteriol 182:6673–6678. https://doi.org/10.1128/JB.182.23.6673-6678.2000
Porter SW, West AH (2003) Ssk1 response regulator binding surface on histidine-containing phosphotransfer protein Ypd1p. Eukaryot Cell 2:27–33. https://doi.org/10.1128/EC.2.1.27-33.2003
Porter SW, West AH (2005) A common docking site for response regulators on the yeast phosphorelay protein YPD1. Biochim Biophys Acta 1748:138–145. https://doi.org/10.1016/j.bbapap.2004.12.009
Janiak-Spens F, Cook PF, West AH (2005) Kinetic analysis of YPD1-dependent phosphotransfer reactions in the yeast osmoregulatory phosphorelay system. Biochemistry 44(1):377–386. https://doi.org/10.1021/bi048433s
Grimshaw CE, Huang S, Hanstein CG, Strauch MA, Burbulys D, Wang L, Hoch JA, Whiteley JM (1998) Synergistic kinetic interactions between components of the phosphorelay controlling sporulation in Bacillus subtilis. Biochemistry 37:1365–1375. https://doi.org/10.1021/bi971917m
Stewart RC (1997) Kinetic characterization of phosphotransfer between CheA and CheY in the bacterial chemotaxis signal transduction pathway. Biochemistry 36:2030–2040. https://doi.org/10.1021/bi962261k
Acknowledgments
This work is part of the Doctoral Thesis of NCM and the sabbatical leave/ENCB-IPN of AVRT. N.C.M. is a CONACyT and BEIFI fellow. This work was supported by the “Secretaria de Investigación y Posgrado IPN” under grants: SIP 20161552, 20161239 and SIP20171476. J.A.C.V. and A.V.R.-T. are COFAA-IPN (COMISIÓN DE OPERACIÓN Y FOMENTO DE ACTIVIDADES ACADÉMICAS DEL INSTITUTO POLITÉCNICO NACIONAL), EDI-IPN (ESTIMULOS AL DESEMPEÑO DE LOS INVESTIGADORES DEL INSTITUTO POLITÉCNICO NACIONAL) and SNI-CONACyT (SISTEMA NACIONAL DE INVESTIGADORES DEL CONSEJO NACIONAL DE CIENCIA Y TECNOLOGÍA) fellows, respectively. N.O.P. is a SNI-CONACyT fellow. The English was edited by the Edanz Editing Sevices.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
CMN_01
Table showing the DOPE, RMSD, QMEAN, and DFire-Energy values of the five models with lowest DOPE of 20 obtained with MODELER 9.14 for CgYpd1 and CgSln1p (RR). The best models are marked in gray. (DOCX 15 kb)
CMN_02
In silico protein structure of the best CgYpd1 model in protein data bank (pdb) format. (PDB 95 kb)
CMN_03
Ramachandran plot analysis of model CgYpd1p selected to perform the molecular dynamics. The analysis confirms the quality in high resolution of the protein structure because most of the φ and ψ bonds are in the allowed zones. (PDF 13 kb)
CMN_04
In silico protein structure of the best CgSln1-RR model in pdb format. (PDB 76 kb)
CMN_05
Ramachandran plot analysis of model CgSln1-RR selected to perform the molecular dynamics. The analysis confirms the quality in high resolution of the protein structure because most of the φ and ψ bonds are in the allowed zones. (PDF 7 kb)
CMN_06
Graphical representation, using VMD software, of the RMSD trajectory of the molecular dynamics of the CgSln1RR-CgYpd1p complex during 100 ns in the absence of a phosphate group. (GIF 273 kb)
CMN_07
Graphical representation, using the VMD, of the RMSD trajectory of the molecular dynamics of the CgSln1RR-CgYpd1p complex during 100 ns in the presence of a phosphate group. (GIF 278 kb)
CMN_08
Video (mpg) of the molecular dynamics between the CgSln1RR-CgYpd1p complex during 100 ns in the absence of the phosphate group. The video shows an unstable interaction between the Asp58 of CgSln1p and His63 of CgYpd1p. (MPG 10121 kb)
CMN_09
Video (mpg) of the molecular dynamics between the CgSln1RR-CgYpd1p complex during 100 ns in the presence of the phosphate group and Mg++ as stabilizer. The video shows a clear and stable interaction between the Asp58 of CgSln1p and His63 of CgYpd1p. (MPG 11497 kb)
Rights and permissions
About this article

Cite this article
Carapia-Minero, N., Castelán-Vega, J.A., Pérez, N.O. et al. The phosphorelay signal transduction system in Candida glabrata: an in silico analysis. J Mol Model 24, 13 (2018). https://doi.org/10.1007/s00894-017-3545-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-017-3545-z