
Abstract
The Pd-catalyzed dearomatization of naphthalene allyl chloride with allyltributylstannane has been investigated using density functional theory (DFT) calculations at the B3LYP level. The calculations indicate that the (ŋ 1-allyl)(ŋ 3-allyl)Pd(PH3) complex is responsible for the formation of ortho-dearomatized product. Moreover it is easy to produce the ortho-dearomatized product when reductive elimination starts from (ŋ 3-allylnaphthalene)(ŋ 1-allyl)Pd complex 7, while it is easy to form the para-dearomatized product when reductive elimination starts from (ŋ 3-allylnaphthalene)(ŋ 1-allyl)Pd complex 9. The Stille coupling products can’t be produced due to high reaction energy barrier.

Two mechanisms of dearomatization are investigated by DFT, and (ŋ 1-allyl)(ŋ 3-allyl)Pd(PH3) complexes are the main intermediates for ortho-dearomatized product.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The alicyclic complexes frequently appear in the molecules of natural products and bioactive compounds. Therefore the synthesis of alicyclic compounds has received considerable interest in recent years. Aromatic compounds are stable and widely available. Thus it is an important method to synthesize alicyclic complexes by dearomatization reaction [1–7]. However, the dearomatization of arenes is especially difficult because the delocalization of π bonds makes aromatic compounds very stable. Over the past few decades, many methods have been developed to accomplish the dearomatization reaction, such as electrophilic addition [8], nucleophilic addition [9, 10], photocycloaddition [11], and so on [12]. Remarkably, the transition-metal-mediated dearomatization methodology has been an efficient method, due to the complexation of aromatic system to transition metals that leads to the activation of arenes and facilitates the electrophilic addition of [M(η 2-arene)] (M = Os, Re, Mo, and W) complexes and the nucleophilic addition of [M(η 6-arene)] (M = Cr, Mn, and Ru) complexes [13].

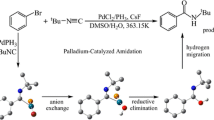
Recently, Bao and co-workers [14] reported the dearomatization reaction of naphthalene allyl chloride with allyltributylstannane in the presence of Pd catalyst, which is displayed in Eq. (1). The ortho-dearomatized product, not the para-dearomatized and Stille cross-coupling products, is synthesized. Meanwhile, they proposed a plausible mechanism (Scheme 1). In the past few years, there have been many DFT studies on mechanisms of similar reactions. For example, Ariafard and co-workers [15] studied the Pd-catalyzed para-dearomatization reaction of benzyl chloride with allyltributylstannane. Ren and co-workers [16] reported the Pd-catalyzed dearomatization of naphthalene allyl chloride with allenyltributylstannane, which also produce the ortho-dearomatized product. Then Ren and co-workers [17] investigated a theoretical study on the mechanism of palladium-catalyzed para-dearomatization of chloromethylnaphthalene with allenyltributylstannane. However, no theoretical study has been reported about the ortho-dearomatization of naphthalene allyl chloride with allyltributylstannane in the presence of Pd catalyst. Therefore many details are lacking for the reaction mechnism. For example, what are the structures of transition states? Why does this reaction form ortho-dearomatized product, not para-dearomatized product? Why does this reaction not give Stille cross-coupling product?

The mechanism proposed by Bao and co-workers
To answer the questions raised above and provide insight into portions of this unusual reaction, we have studied the Pd-catalyzed dearomatization of naphthalene allyl chloride with allyltributylstannane using the B3LYP density functional theory. We investigate the mechanism proposed by Bao, in which the main intermediates are bis(ŋ 3-ally)palladium complexes. Then a different mechanism is calculated, in which the main intermediates are (ŋ 1-allyl)(ŋ 3-allyl)Pd(PH3) complexes. In addition to the ortho-dearomatized product, the pathways to generate the para-dearomatized and Stille cross-coupling products are also examined.
Computational details
All calculations in this study are performed with Guassian03 suite of programs [18]. The geometries of the reactants, transition states, intermediates, and products are fully optimized without any symmetry constraints at the B3LYP level [19–21] of density functional theory (DFT). Frequencies are computed at the same level of theory for all the stationary points to characterize the transition states (one imaginary frequency) and the equilibrium structures (no imaginary frequency). The effective core potentials of Hay and Wadt with double-ξ valance basis sets (LanL2DZ) [22, 23] are used for Pd, Sn, and P; the standard polarized 6-31G(d) basis set is chosen to describe C and Cl; and H is described by 6-31G basis set [24, 25]. Polarization functions are added for Sn(ξd = 0.183), C(ξd = 0.8), P(ξd = 0.34), and Cl(ξd = 0.514) [26]. Calculations of intrinsic reaction coordinates (IRC) [27, 28] are performed on transition states to confirm that such structures are indeed connecting two minima. Natural charges and electron occupancy are analyzed with natural bond orbital (NBO) [29]. The triphenylphosphine ligand PPh3 is modeled by PH3, the geometry around the metal center in each of the structures is only a little different from the structures that were calculated with the triphenylphosphine [15, 16].
Results and discussion
Dearomatization reaction mechanisms
Two kinds of dearomatization reaction mechanisms for ortho-dearomatized products are examined. As shown in Scheme 2, the main intermediates are bis(ŋ 3-ally)palladium complexes in mechanism 1 reported by Bao and co-workers [14]. In Scheme 4, the main intermediates are (ŋ 1-allyl)(ŋ 3-allyl)palladium complexes in mechanism 2.

Mechanism 1
As shown in mechanism 1(Scheme 2), the (ŋ 3-ally)(ŋ 3-allylnaphthalene)palladium intermediate 3 isomerizes to (ŋ 3-ally)(ŋ 3-allylnaphthalene)palladium complex 4 via TS3-4 that completes the rearrangement of bis(ŋ 3-ally)palladium complex. Reductive elimination step starts from intermediate 4 by directly coupling the C of ŋ 3-ally ligand with the ortho carbon of the ŋ 3-allylnaphthalene ligand via TS4-5.
In mechanism 1, because the coordination mode of intermediate 3 is bis(ŋ 3-ally)palladium, the pathway includes the cis-structure and trans-structure pathways starting from intermediate 3. The cis-structure and trans-structure are shown in Scheme 3. Figure 1 shows the energy profiles of the cis-structure and trans-structure pathways. As shown in Fig. 1, the trans-intermediates (3trans, 4trans, and 5trans) are more stable than the cis-intermediates (3cis, 4cis, and 5cis). However, the cis-transition states have relatively lower energy than the trans-transition states. The energy barriers of cis-structure pathway and trans-structure pathway are 44.28 and 53.73 kcal mol-1, respectively. The cis-structure pathway is more favorable kinetically than trans-structure pathway. Therefore, mechanism 1 is not feasible kinetically because of a relatively large energy barrier.

cis-structure (left) and trans-structure (right)

Relative energy profiles for mechanism 1 (dotted and solid lines are the trans-structure and cis-structure pathways, respectively)
Because of the relatively large energy barrier in mechanism 1, mechanism 2 is proposed as shown in Scheme 4. The (ŋ 1-allylnaphthalene)(ŋ 3-allyl)Pd(PH3) intermediate 6 isomerizes to (ŋ 3-allylnaphthalene)(ŋ 1-allyl)Pd(PH3) intermediate 7 via TS6-7 that completes the rearrangement of (ŋ 1-allyl)(ŋ 3-allyl)Pd(PH3) complex. Reductive elimination step starts from intermediate 7 by directly coupling terminal carbon of the ŋ 1-ally ligand with the ortho carbon of ŋ 3-allylnaphthalene ligand via TS7-8.

Mechanism 2
In mechanism 2, because the coordination mode of TS6-7 is bis(ŋ 3-ally)palladium, the pathway includes the cis-structure and trans-structure pathways starting from TS6-7. As shown in Fig. 2, the energy barriers of the cis-structure and trans-structure pathways are 18.68 and 24.21 kcal mol-1, respectively. The cis-structure pathway is more favorable kinetically than trans-structure pathway. Compared to mechanism 1, the energy barriers of mechanism 2 are lower by respective 25.60 (cis-structure pathway) and 29.52 (trans-structure pathways) kcal mol-1. Therefore mechanism 2 is more favorable kinetically.

Relative energy profiles for mechanism 2. (dotted and solid lines are the trans-structure and cis-structure pathways, respectively)
Ren and co-workers [16] reported the Pd-catalyzed dearomatization of naphthalene allyl chloride with allenyltributylstannane, which is displayed in Eq. (2). The difference from the present study is taking allenyltributylstannane as the reagent in place of allyltributylstannane. Two kinds of dearomatization reactions in the reductive elimination are compared in Scheme 5.

Comparison of allyl (a) and allenyl (b) ligands in the reductive elimination

As shown in Scheme 5, the intermediates have the same coordination mode in the reductive elimination. Moreover, both of the ŋ 1-ligands have an available electron pair to attack the ortho carbon of ŋ 3-allylnaphthalene ligand via the transition states to form the dearomatization product. For the allyl ligand (Scheme 5a), the reductive elimination takes place from (ŋ 1-allylnaphthalene)(ŋ 3- allyl)Pd(PH3) intermediate 6. Then the intermediate 6 isomerizes to (ŋ 3-allylnaphthalene)(ŋ 1- allyl)Pd(PH3) intermediate 7. The dearomatization product is formed by directly coupling terminal carbon of the ŋ 1-ally ligand with the ortho carbon of ŋ 3-allylnaphthalene ligand via TS7-8. For the allenyl ligand (Scheme 5b), the reductive elimination takes place from (ŋ 1-allylnaphthalene)(ŋ 3-propargyl)Pd(PH3) intermediate 15. Then the intermediate 15 isomerizes to (ŋ 3-allylnaphthalene)(ŋ 1-allenyl)Pd(PH3) intermediate 16. The dearomatization product is formed by directly coupling the terminal carbon of the ŋ 1-allenyl ligand with ortho carbon of the ŋ 3-naphthalene ligand via TS16-17.
Structure analysis
The optimized structural parameters of transition states and intermediates in mechanism 1 and mechanism 2 are shown in Fig. 3. During the rearrangement of bis(ŋ 3-ally)palladium complex in mechanism 1, the Pd-C5, Pd-C6, and Pd-C7 bonds in TS3-4 are 2.88, 2.11, and 3.07 Å. During the rearrangement of (ŋ 1-allyl)(ŋ 3-allyl)Pd(PH3) complex in mechanism 2, the Pd-C4, Pd-C5, and Pd-C6 bonds in TS6-7 are 2.21, 2.19, and 2.40 Å. Therefore there is a stronger Pd- ŋ 3-allylnaphthalene bond interaction in TS6-7 than in TS3-4. During the reductive elimination, there is a larger conjugated π system in TS7-8 than TS4-5.

Optimized structural parameters of transition states in mechanism 1 and mechanism 2 (bond lengths are in Å)
The stability of transition states of mechanism 1 and 2 are investigated by NBO method. The NBO charge of TS3-4, TS4-5, TS6-7, and TS7-8 are summarized in Table 1. In TS3-4 and TS6-7, the positive charges of Pd are respectively 0.447e and 0.517e, while the negative charges of the sum of C1-C8 are −2.729e and −2.909e, respectively. In TS4-5 and TS7-8, the positive charges of Pd are respectively 0.248e and 0.325e, while the negative charges of the sum of C1-C8 are −2.558e and −2.808e, respectively. There is more electron transfer from Pd to coordinated C atoms in TS6-7 and TS7-8 than in TS3-4 and TS4-5. Furthermore, in reductive elimination, PH3 donates an electron to Pd in mechanism 2.
The orbital occupancies of 5s and 4d of Pd in TS3-4, 4, TS4-5, TS6-7, 7, and TS7-8 are listed in Table 2. The orbital occupancies of 4d orbital of Pd in TS3-4 (9.232e) and in TS4-5 (9.433e) are larger than the ones in TS6-7 (9.219e) and TS7-8 (9.359), respectively. The orbital occupancies of 5s and 4d of Pd are consistent with the previous charge analysis of Pd in TS3-4, TS4-5, TS6-7, and TS7-8.
As shown in Table 2, from intermediate 7 to TS7-8, the orbital occupancies of 5s orbital decrease from 0.358e to 0.305e, while the orbital occupancies of 4d orbital increase from 9.243e to 9.359e. In TS7-8, the electrons transfer from the π orbital of allyl to 4d orbital of Pd, with the backdonation from 5s orbital of Pd to π* orbital of allyl. Therefore, the backdonation interaction makes the TS7-8 more stable. From the geometric and electronic properties, TS6-7 and TS7-8 are more stable than TS3-4 and TS 4–5, respectively. Thus mechanism 2 is more favorable than mechanism 1.
Possible pathway to para-dearomatized product
The possible pathway to generate para-dearomatized product is studied. As displayed in Scheme 2, the process from 7 to 8 via TS7-8 is a crucial step for determining the dearomatized product. Therefore we start the calculation from intermediate 7. Firstly, intermediate 7 isomerizes to 9 that the Pd coordinates to the naphthalene ring, forming (ŋ 3-allylnaphthalene)(ŋ 1-allyl)Pd(PH3) intermediate 9, which is shown in Fig. 4. Comparing intermediate 7 (in Fig. 2) and intermediate 9 (in Fig. 4), the intermediate cis-7 and trans-7 are more stable than 9 by 5.30 and 4.15 kcal mol-1, respectively. Then intermediate 9 forms para-dearomatized product 10 via TS9-10, the overall barrier is 14.76 kcal mol-1, which is 3.92 kcal mol-1 lower than the largest energy barrier in mechanism 2. Therefore para-dearomatization reaction is energetically feasible. It is easy to produce the ortho-dearomatized product when reductive elimination starts from (ŋ 3-allylnaphthalene)(ŋ 1-allyl)Pd(PH3) intermediate 7. It is easy to form the para-dearomatized product when reductive elimination starts from (ŋ 3-allylnaphthalene)(ŋ 1-allyl)Pd(PH3) intermediate 9.

Relative energy profiles for the possible dearomatization product from intermediate 9
Possible pathway to Stille coupling product
The reaction is possible to generate Stille coupling products, which are expected products of organic electrophiles (RX) with organostannanes(R′SnR″3) [30]. The pathways directly coupling the allyl and the propenylnaphthalene groups are calculated from the intermediates 3 cis and 6. Figure 5 shows the energy profiles calculated for two direct coupling pathways. The energy barriers of two pathways are calculated to be 42.43 (TS3–11) and 22.44(TS6–14) kcal mol-1, respectively. The energy barriers of direct coupling are higher than the energy barrier of dearomatization (18.68 kcal mol-1). Consistent with the experimental observation, the dearomatized product is produced in palladium-catalyzed dearomatization reaction of propenylnaphthalene chlorine with allyltributylstannane.

Relative energy profiles calculated for the possible Stille coupling product from intermediate 3 cis (left) and 6 (right)
Conclusions
The palladium-catalyzed dearomatization reaction of propenylnaphthalene chlorine with allyltributylstannane has been studied using density functional theory calculations at the B3LYP level. Our calculation shows the energy barrier of mechanism 1 is 44.28 kcal mol-1 (trans-structure pathway is 53.73 kcal mol-1), which is unfavorable kinetically. The energy barrier of mechanism 2 is 18.68 kcal mol-1 (trans-structure pathway is 24.21 kcal mol-1). Therefore this ortho-dearomatization reaction proceeds by mechanism 2, in which (ŋ 1, ŋ 3-allyl) palladium complexes are the main intermediates and reductive elimination starts from (ŋ 3-allylnaphthalene)(ŋ 1-allyl)Pd(PH3) intermediate 7. By the analysis of geometry, NBO charge and orbital occupancies of 5s and 4d of Pd, the transition states in mechanism 2 are more stable than transition states in mechanism 2. Moreover the cis-structure pathway is more favorable than trans-structure pathway. The pathway to para-dearomatization product is examined from (ŋ 3-allylnaphthalene)(ŋ 1-allyl)Pd(PH3) intermediate 9. The energy barrier is only 14.76 kcal mol-1. Thus the para-dearomatized product is easily formed when reductive elimination starts from (ŋ 3-allyl naphthalene)(ŋ 1-allyl)Pd(PH3) intermediate 9. The possible Stille coupling products are also examined. The energy barriers of two pathways are calculated to be respectively 42.43 and 22.44 kcal mol-1. Which are higher than the energy barrier of dearomatization reaction(18.68 kcal mol-1). At last, we illustrate the similarity between allayl and allenyl ligands in the reductive elimination, in which the intermediates have the same coordination mode and both of the ŋ 1-ligands have an available electron pair to interact with the ortho carbon.
References
Bao M, Nakamura H, Yamamoto Y (2001) J Am Chem Soc 123:759–760
García-Fortanet J, Kessler F, Buchwald SL (2009) J Am Chem Soc 131:6676–6677
Lee S, Chataigner I, Piettre SR (2011) Angew Chem Int Ed 50:472–476
Peng B, Feng X, Zhang X, Zhang S, Bao M (2010) J Org Chem 75:2619–2627
Pouységu L, Deffieux D, Quideau S (2010) Tetrahedron 66:2235–2261
Rousseaux S, García-Fortanet J, Del Aguila Sanchez MA, Buchwald SL (2011) J Am Chem Soc 133:9282–9285
Rudolph A, Bos PH, Meetsma A, Minnaard AJ, Feringa BL (2011) Angew Chem Int Ed 50:5834–5838
Delafuente DA, Myers WH, Sabat M, Harman WD (2005) Organometallics 24:1876–1885
Clayden J, Kenworthy MN, Helliwell M (2003) Org Lett 5:831–834
Zhou L, Wu LZ, Zhang LP, Tung CH (2006) Organometallics 25:1707–1711
Kohmoto S, Masu H, Tatsuno C, Kishikawa K, Yamamoto M, Yamaguchi K (2000) J Chem Soc Perkin Trans 1:4464–4468
Boivin J, Yousfi M, Zard SZ (1997) Tetrahedron Lett 38:5985–5988
Pape AR, Kaliappan KP, Kündig EP (2000) Chem Rev 100:2917–2940
Lu S, Xu Z, Bao M, Yamamoto Y (2008) Angew Chem Int Ed 47:4366–4369
Ariafard A, Lin Z (2006) J Am Chem Soc 128:13010–13016
Ren Y, Jia J, Zhang T, Wu H, Liu W (2012) Organometallics 31:1168–1179
Ren Y, Jia J, Liu W, Wu H (2013) Organometallics 32:52–62
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven T Jr, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, and Pople JA (2004) Gaussian Inc, Wallingford
Becke AD (1993) J Chem Phys 98:5648
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785–789
Miehlich B, Savin A, Stoll H, Preuss H (1989) Chem Phys Lett 157:200–206
Hay PJ, Wadt WR (1985) J Chem Phys 82:270
Hay PJ, Wadt WR (1985) J Chem Phys 82:299
Davidson ER, Feller D (1986) Chem Rev 86:61–696
Hariharan PC, Pople JA (1973) Theor Chim Acta 28:213–222
Ehlers AW, Böhme M, Dapprich S, Gobbi A, Höllwarth A, Jonas V, Köhler KF, Stegmann R, Veldkamp A, Frenking G (1993) Chem Phys Lett 208:111–114
Fukui K (1970) J Chem Phys 74:4161–4163
Fukui K (1981) Acc Chem Res 14:363–368
Reed AE, Curtiss LA, Weinhold F (1988) Chem Rev 88:899–926
Milstein D, Stille JK (1979) J Am Chem Soc 101:4992–4998
Acknowledgments
This work was supported by the National Science Foundation of China (21001019) the Fundamental Research Funds for the Central Universities (DUT15LK24). The results were obtained on the ScGrid of Supercomputing Center, Computer Network Information Center of Chinese Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Cao, W., Tian, D. & Han, D. DFT studies on the palladium-catalyzed dearomatization reaction between naphthalene allyl chloride and allyltributylstannane. J Mol Model 21, 260 (2015). https://doi.org/10.1007/s00894-015-2802-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-015-2802-2