
Abstract
Quantum chemical calculations have been per-formed for the complexes of formamidine (FA) and hypohalous acid (HOX, X = F, Cl, Br, I) to study their structures, properties, and competition of hydrogen bonds with halogen bonds. Two types of complexes are formed mainly through a hydrogen bond and a halogen bond, respectively, and the cyclic structure is more stable. For the F, Cl, and Br complexes, the hydrogen-bonded one is more stable than the halogen-bonded one, while the halogen-bonded structure is favorable for the I complexes. The associated H-O and X-O bonds are elongated and exhibit a red shift, whereas the distant ones are contracted and display a blue shift. The strength of hydrogen and halogen bonds is affected by F and Li substitutents and it was found that the latter tends to smooth differences in the strength of both types of interactions. The structures, properties, and interaction nature in these complexes have been understood with natural bond orbital (NBO) and atoms in molecules (AIM) theories.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Intermolecular interactions have been attracting much attention due to their extensive applications in fields of chemistry, biology, and physics [1, 2]. Hydrogen bond (HB) is one of the most important intermolecular interactions and its formation is mainly due to electrostatic interaction, together with induction and dispersion interactions [3, 4]. Halogen bond (XB) is another important intermolecular interaction and recently more attention has been paid to XB because it exhibits similar applications to HB [5–10]. The origin of XB formation is dependent on the nature of halogen donor and acceptor [11–15]. The above applications of HB and XB are related to their directionality and strength. The directionality of XB is often attributed to anisotropic distribution of electrostatic potentials on the covalent halogen atomic surface [16]. Very recently, Stone studied some simple halogen-bonded complexes using symmetry-adapted perturbation theory (SAPT) and concluded that electrostatics are not single decisive factor for their geometries and the strong tendency to linearity of XB is a consequence of exchange-repulsion in some particular cases [17].
Besides the atoms having lone pair electrons such as O and N, the halogen acceptors in XBs include π systems, metal hydrides, radicals, and carbenes [18–22]. The strength of XB is also dependent on the nature of halogen atom. Fluorine seldom participates in XB, although it sometimes does when it combines with strong Lewis bases or strong electron-withdrawing groups adjoin with it [23]. The strength of XB increases as follows: F < Cl < Br < I, and iodine forms the strongest XB. In general, XB is comparable to HB in strength, thus they show competition when the hydrogen and halogen donors exist simultaneously [24]. A versatile strategy for the assembly of discrete supermolecules and heteromeric molecular architectures was suggested by means of structural competition between HBs and XBs [25].
Hypohalous acids play an important role both in atmospheric chemistry involved in catalytic cycles in the seasonal depletion of the ozone layer in the stratosphere [26–28] and in pathophysiological processes as oxidants with potent antibacterial properties [29] as well as in the immune defense of mammalians [30]. Because of chemical instability of the hypohalous acids, it is a challenge to probe and quantify them [31, 32]. The coexistence of H and X atoms in hypohalous acids makes them act as the proton and halogen donors in HB and XB. Both types of interactions are of importance for the understanding of the above chemical and biological processes. More importantly, they provide a good model to study the competition of HB with XB [33–45]. In general, the hydrogen-bonded complexes are more stable for HOCl and HOBr, while the halogen-bonded isomers are more stable for HOI. In addition, the halogen acceptor and substituents have an effect on their stability. H2CS forms a stronger halogen bond with HOBr than the hydrogen bond [41]. Some studies paid attention to dimers of hypohalous acids [46–48]. Theoretical calculations play a prominent part in unveiling the structures, properties, and nature of these complexes involved with hypohalous acids.
People are interested in amidines because they exhibit a unique and fascinating biological activity, including bactericidal and antiprotozoal effects, antihelminthic, fungicidal and herbicidal properties, and the insecticidal and acaricidal actions [49, 50]. These functions may be related with proton transfer occurring in complexes of amidines with itself [51], formic acid [52], formamide [53], glycinamide [54], water [55], and alcohols [56].
Because of the important role of amidines and hypohalous acids in both atmospheric and biological processes, there is an interest in studying the interactions between them. In the present paper, the complexes of formamidine (FA) with hypohalous acids HOX (X = F, Cl, Br, I) have been investigated to study the competition between HB and XB in these complexes. To the best of our knowledge, neither theoretical nor experimental data regarding the structural information for these complexes are available in the literature. The present work presents a detailed examination of the stabilities, electronic structure, and vibrational frequencies of these complexes.
Theoretical methods
The structures have been optimized at the MP2 computational level with the aug-cc-pVTZ basis set for all atoms except I atom, for it the aug-cc-pVTZ-PP basis set was adopted. Frequency calculations have been carried out at the same level to confirm that the structures obtained correspond to energetic minima. The interaction energy was calculated with supermolecular method by subtracting the energy sum of the isolated monomers from the energy of the complex. The interaction energy with such method is susceptible to basis-set superposition error (BSSE), which can be removed with the counterpoise method suggested by Boys and Bernardi [57]. All calculations were performed using the Gaussian 09 package of codes [58].
The natural bond orbital (NBO) method [59] implemented within the Gaussian 09 program has been used to analyze the interaction of occupied and empty orbitals as well as charge transfer in these complexes. The AIM2000 package [60] was used to obtain bond properties in view of topology including electron density, Laplacian, and energy density. The electrostatic potentials at the 0.001 electrons Bohr-3 isodensity surfaces were calculated with the Wave Function Analysis-Surface Analysis Suite (WFA-SAS) program [61].
Results and discussion
FA-(Z)-HOX systems
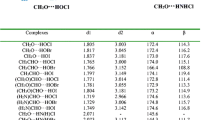
Figure 1 shows the structures of FA-(Z)-HOX and FA-(Z)-XOH (X = F, Cl, Br, I) complexes. In FA-(Z)-HOX, two types of HBs coexist. One is OH···N HB with the imino nitrogen atom as the proton acceptor and HOX as the proton donor, the other is CH···X HB, whith the halogen as the proton acceptor and the C-H proton as the donor. In FA-(Z)-XOH, there is an OX···N XB with the imino nitrogen atom as the halogen acceptor and HOX as the halogen donor, together with a similar CH···X contact to that in FA-(Z)-HOX. It has been demonstrated that HB and XB are electrostatically-driven [11, 16, 62, 63], thus the formation of HB and XB in the FA complexes with HOX can be understood with the electrostatic potentials of FA and HOX, as shown in Fig. 2. The positive region of electrostatic potentials on the H and X atoms in HOX points to the negative region of electrostatic potentials on the N atom in FA. Simultaneously, the negative region of electrostatic potentials on the X atom in HOX is close to the negative region of electrostatic potentials on the H atom in FA. The OH···N HB and OX···N XB are dominant in the corresponding isomers, while the CH···X contact is a secondary one. The respective binding distances are summarized in Table 1. The H···N distance is 1.71–1.73 Å, X···N distance is 2.44–2.55 Å, and the H···X distance is 2.55–3.26 Å. The values of H···N and X···N distances are smaller than the sum of the van der Waals radii of the respective atoms (2.6 Å for H and N atoms, 3.3 Å for Cl and N atoms, 3.5 Å for Br and N atoms, 3.6 Å for I and N atoms). This supports the presence of the OH···N HB in FA-(Z)-HOX and the OX···N XB in FA-(Z)-XOH. The H···X distance almost amounts to or larger than the sum of the van der Waals radii of the respective atoms (2.5 Å for H and F atoms, 2.9 Å for H and Cl atoms, 3.1 Å for H and Br atoms, 3.3 Å for H and I atoms). This shows that the H···X interaction is very weak in both types of isomers. These interactions are confirmed by the presence of intermolecular bond critical points (BCPs) (red small points in Fig. 3). In FA-(Z)-HOX, the combination of the main interaction with the secondary one results in formation of a ring structure. This can be confirmed with the presence of a ring critical point (RCP) in the structure (a yellow small point in Fig. 3).

The structures of FA-(Z)-HOX and FA-(E)-HOX (X = F, Cl, Br, I) systems

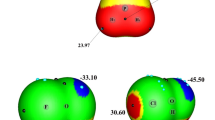
The electrostatic potentials of FA and HOX calculated at the MP2/aug-cc-pVTZ level. Color ranges, in kcal mol−1, are: red, greater than 18.8; yellow, between 18.8 and 0; green, between 0 and −25.1; blue, less than −25.1

Molecular graphs of FA-(Z)-HOX and FA-(E)-HOX (X = F, Cl, Br, I) systems. Small red balls indicate the bond critical points and yellow ones show the ring critical point
The interaction energy is a convincing measurement for the stability of complexes. For the HOCl and HOBr complexes, FA-(Z)-HOX is more stable than FA-(Z)-XOH, while a reverse result is found for the HOI complexes. Interestingly, the interaction energies are almost equal for FA-(Z)-HOF, FA-(Z)-HOCl, and FA-(Z)-HOBr complexes, although three halogen atoms have different electronegativity. The greater electronegativity of F in HOF should make the proton form a stronger HB than HOCl and HOBr. However, the H···N distance in FA-(Z)-HOF is larger than in the respective HOCl and HOBr systems. This abnormal result has been demonstrated in the hydrogen-bonded complexes of hypohalous acids with nitrogenated bases [39]. This may be attributed to the nature of O-X bond in HOX. The O–F bond is a covalent, polarized bond, while other O-X bond is of electron donor–acceptor-type with the halogen donating electron density to the valence shell of oxygen [64]. For FA-(Z)-HOX (X = Cl, Br, I), with the increase of the halogen atom mass the interaction energy becomes less negative, the H···N distance is bigger, and the H···X distance is also increased. This agrees with the change of the heavier halogen atom electronegativity and its atom radius. For FA-(Z)-XOH, with the increase of the halogen atom mass the interaction energy becomes more negative, and this result is consistent with the most positive electrostatic potential on the halogen atomic surface in HOX [39]. This indicates that the electrostatic interaction is very important in the formation of halogen bond. An interesting result is found for the smallest Br···N distance, although the interaction strength is not largest and the Br atomic radius is not smallest. The H···X distance in FA-(Z)-XOH is greater than that in FA-(Z)-HOX, indicating a much weaker contact in the former.
Upon complexation, the H-O bond is elongated in FA-(Z)-HOX and shortened in FA-(Z)-XOH, whereas the X-O bond is lengthened in FA-(Z)-XOH and contracted in FA-(Z)-HOX except in FA-(Z)-HOF. The value of the bond elongation is bigger than that of the bond contraction. Accompanied with the bond elongation and contraction, the respective bond stretch vibrations exhibit a red shift and blue shift except for the F-O bond in FA-(Z)-HOF. The red shift is more prominent for the H-O bond than for the X-O bond owing to the lighter mass of H atom.
In nearly all of the cases there are simultaneous interactions that contribute to the computed total interaction energies. Thus the orbital interaction between the orbital donor and the acceptor is used to understand the formation of the complexes, although it is not observed physically [65]. The analyzed orbital interactions are LPN→BD*O-H and LPX→BD*C-H in FA-(Z)-HOX, while they are LPN→BD*O-X and LPX→BD*C-H in FA-(Z)-XOH. These orbital interactions are estimated with the second-order perturbation energy, and the corresponding results are given in Table 2. The LPX→BD*C-H orbital interaction is far smaller than LPN→BD*O-H and LPN→BD*O-X ones, showing the secondary interaction can be negligible. The LPX→BD*C-H orbital interaction in FA-(Z)-HOX is larger than that in FA-(Z)-XOH, and this is in agreement with the H···X distance in both types of complexes. The perturbation energies due to the LPN→BD*O-H and LPN→BD*O-X orbital interactions have a consistent change with the interaction energy in both types of complexes, indicating the orbital interaction has a contribution to the formation of the complexes. For the Cl and Br complexes, the orbital interaction is stronger in FA-(Z)-HOX than that in FA-(Z)-XOH, however it is stronger in FA-(Z)-IOH. This means that the competition between HB and XB can be regulated through the change of halogen donor.
Accompanied with the above orbital interactions, charge transfer occurs from the electron donor to the acceptor. With the increase of the halogen atomic number, the charge transfer decreases in FA-(Z)-HOX (X = Cl, Br, I) but increases in FA-(Z)-XOH. A good relationship is found for the charge transfer and the interaction energy for the Cl, Br, and I complexes. These orbital interactions lead to the change of electron densities in the H-O and O-X sigma bonding and sigma antibonding orbitals. One can see from Table 2 that the electron densities in the sigma bonding orbitals have a small decrease, while they show a great increase for the σ*H-O in FA-(Z)-HOX and for the σ*X-O in FA-(Z)-XOH. This increase is responsible for the elongation and red shift of H-O and X-O bonds.
The theory of atoms in molecules (AIM) is another means for judging the formation of complexes, estimating their strength, and unveiling the nature of interactions. For these goals, in Table 3 we analyzed these complexes with the topological parameters including the electron density (ρ), Laplacian (∇2ρ), and energy density (H). For the H···N HB and X···N XB, the ρ values range from 0.03 au to 0.05 au, while the ∇2ρ ones are in range of 0.08–0.13 au. The latter conforms to the range (0.02–0.15 au) suggested for HBs by Koch and Popelier [66], while the former in most complexes is out of the range (0.002–0.04 au) [66]. The ρ value has a consistent change with the interaction energy in both types of complexes and it can be used to estimate the interaction strength [67]. The greater ρ values mean stronger interactions in these complexes, which are confirmed by bigger interaction energies. The strong interactions in these complexes are also evidenced by the negative H value because the latter corresponds to partly covalent interactions [68]. An exception is found in FA-(Z)-ClOH, in which the electron density is small and the energy density is positive. For the H···X BCP in FA-(Z)-HOX, the electron density is much smaller than those for the H···N HB and X···N XB and the energy density is positive, corresponding to the weak interaction. The electron density at the H···X BCP in FA-(Z)-HOX (X = Cl, Br, I) has an increase for the heavier halogen complex, confirming the change of H···X HB strength. According to the formulas of E HB = 1/2V [69], where V is potential energy density, the interaction energies of H···N and H···Cl HBs are calculated to be −15.8 and −0.99 kcal mol−1, respectively, in FA-(Z)-HOCl. This further shows that the contribution from the H···X HB is very small in FA-(Z)-HOX.
FA-(E)-HOX systems
Figure 1 also shows the structures of FA-(E)-HOX and FA-(E)-XOH (X = F, Cl, Br, I) complexes. The similar H···N HB and X···N XB are found for FA-(E)-HOX and FA-(E)-XOH, respectively. The difference is the NH···X HB formed between the proton of amino and the halogen atom. The existence of both types of interactions in FA-(E)-HOX and FA-(E)-XOH as well as the formation of cyclic complexes with them are evidenced with the BCPs and RCP respectively in Fig. 3. Clearly, the FA-(E)-HOX system is more stable than the FA-(Z)-HOX counterpart. The difference of the interaction energy between FA-(E)-HOX and FA-(Z)-HOX systems becomes smaller for the hydrogen-bonded complex but larger for the halogen-bonded complex with the increase of the halogen atom mass. The H···N and X···N distances in FA-(E)-HOX system are shorter than those in the FA-(Z)-HOX counterpart. The H···X distance in FA-(E)-HOX system shows a rise tendency with the increase of the halogen atom mass, and it is smaller in FA-(E)-HOX than in FA-(E)-XOH, showing a stronger H···X HB in the former. The H-O and X-O bond lengths as well as the respective frequency shifts in FA-(E)-HOX system show similar changes to those in FA-(Z)-HOX system.
Similar orbital interactions are analyzed for the H···N HB and X···N XB in FA-(E)-HOX system, except the LPX→BD*N-H orbital interaction for the NH···X HB. The second-order perturbation energies due to the LPN→BD*O-H and LPN→BD*O-X orbital interactions have a similar change as shown in the above discussion, although they are much larger in FA-(E)-HOX system. The LPX→BD*N-H orbital interaction of the secondary interaction in FA-(E)-HOX system is stronger than the LPX→BD*C-H orbital interaction of the secondary interaction in FA-(Z)-HOX system and it is stronger in FA-(E)-HOX than in FA-(E)-XOH. In both types of complexes, however, it is stronger for the heavier halogen atom. This is inconsistent with the halogen electronegativity and means the electrostatic interaction plays a minor role in the NH···X HB. Again, the electron density is increased in the σ*H-O and σ*X-O orbitals upon complexation.
The bigger electron densities at the H···N and X···N BCPs indicate the stronger HB and XB interactions in FA-(E)-HOX system. The partly covalent nature of H···N HB and X···N XB is confirmed with the negative H value in the complexes except in FA-(E)-ClOH. The NH···X HB corresponds to the smaller electron density and the positive H value at the H···X BCP.
Effect of Li and F substitutents
The above results show that HB is stronger than XB in the Br complexes. It has been demonstrated that the strength of HB and XB can be regulated by substituents [41]. Thus the H atom of C-H bond in FA is replaced with F and Li substitutents. The results of the corresponding Br complexes are presented in Table 4. The electron-withdrawing F group weakens the HB and XB interactions, while the electron-donating Li group strengthens them. The former brings out a similar weakening effect on the HB and XB, while the latter causes a greater enhancement on the XB. The difference in the interaction energy of HB and XB decreases from 3.5 kcal mol−1 for the unsubstituted complex to 1.3 kcal mol−1 for the Li-substituted one. The geometric and spectroscopic parameters undergone a corresponding change accompanied by the change in the strength of HB and XB. An abnormal result is that the H···Br distance becomes shorter due to the Li substitution. Surprisingly, a similar substitution makes the O-Br bond have a prominent elongation (0.122 Å). A very big red shift (−1280 cm-1) is found for the H-O stretch vibration in Li-FA-(E)-HOBr. Although the Li substitution does not make XB stronger than HB, it decreases their difference in strength.
Conclusions
The complexes of formamidine and hypohalous acid (HOX, X = F, Cl, Br, I) have been investigated at the MP2/aug-cc-pVTZ level. Formamidine is not only a good proton acceptor but a good halogen acceptor as well. The hydrogen-bonded complexes are more stable for X = F, Cl, Br, while the halogen-bonded complexes are favorable for X = I. Their stability can be regulated by substitution effect. Both molecules have biological activity, thus the study on the interaction between them is interesting. This work is helpful for understanding the mechanism behind their biological activity.
References
Scheiner S (1997) Hydrogen bonding: a theoretical perspecitive. Oxford University Press, New York
Jeffrey GA (1997) An introduction to hydrogen bonding. Oxford University Press, New York
Czyznikowska Z (2009) J Mol Struct: THEOCHEM 895:161–167
Scheiner S (2013) Int J Quantum Chem 113:1609–1620
Gilday LC, Lang T, Caballero A, Costa PJ, Felix V, Beer PD (2013) Angew Chem, Int Ed 52:4356–4360
Jentzsch AV, Matile S (2013) J Am Chem Soc 135:5302–5303
Khavasi HR, Tehrani AA (2013) Inorg Chem 52:2891–2905
Ormond-Prout JE, Smart P, Brammer L (2012) Cryst Growth Des 12:205–216
El-Sheshtawy HS, Bassil BS, Assaf KI, Kortz U, Nau WM (2012) J Am Chem Soc 134:19935–19941
Meazza L, Foster JA, Fucke K, Metrangolo P, Resnati G, Steed JW (2013) Nat Chem 5:42–47
Politzer P, Murray JS, Clark T (2010) Phys Chem Chem Phys 12:7748–7757
Del Bene JE, Alkorta I, Elguero J (2010) J Phys Chem A 114:12958–12962
Palusiak M (2010) J Mol Struct: THEOCHEM 945:89–92
Zou JW, Jiang YJ, Guo M, Hu GX, Zhang B, Liu HC, Yu QS (2005) Chem Eur J 11:740–751
Tian WK, Miao Q, Li QZ, Li WZ, Cheng JB (2013) Comput Theor Chem 1012:41–46
Clark T, Hennemann M, Murray JS, Politzer P (2007) J Mol Model 13:291–296
Stone AJ (2013) J Am Chem Soc 135:7005–7009
Tomura M (2009) Chem Phys 359:126–131
Raghavendra B, Arunan E (2007) J Phys Chem A 111:9699–9706
Chudzinski MG, McClary CA, Taylor MS (2011) J Am Chem Soc 133:10559–10567
Lipkowski P, Grabowski SJ, Leszczynski J (2006) J Phys Chem A 110:10296–10302
Li QZ, Wang YL, Liu ZB, Li WZ, Cheng JB, Gong BA, Sun JZ (2009) Chem Phys Lett 469:48–51
Metrangolo P, Murray JS, Pilati T, Politzer P, Resnati G, Terraneo G (2011) CrystEngComm 13:6593–6596
Bouchmella K, Boury B, Dutremez SG, van der Lee A (2007) Chem Eur J 13:6130–6138
Aakeroy CB, Fasulo M, Schultheiss N, Desper J, Moore C (2007) J Am Chem Soc 129:13772–13773
Francisco JS, Sander SP (1993) J Chem Phys 99:6219–6220
Molina MJ, Rowland FS (1974) Nature 249:810–812
Rowland FS, Molina MJ (1975) Rev Geophys Space Phys 13:1–35
Thomas E (1979) Infect Immun 23:522–531
Roos D, Winterbourn CC (2002) Science 296:669–671
Yang YC, Lu HH, Wang WT, Liau I (2011) Anal Chem 83:8267–8272
Cheng XH, Jia HZ, Long T, Feng J, Qin JG, Li Z (2011) Chem Commun 47:11978–11980
Solimannejad M, Alkorta I (2008) Chem Phys Lett 454:201–206
Yuan K, Liu YZ, Zhu YC, Zhang J, Zhang JY (2009) Acta Chin Sin 67:499–506
Yuan K, Liu YZ, Lue LL, Ma WC (2008) Acta Chin Sin 24:1257–1263
Yuan K, Liu YZ, Ma WC, Tang HA, Zhu YC, Zhang J (2009) Chi J Chem 27:900–906
Li ZF, Wang XY, Tang HA, Zhu YC, Li HY (2009) Chem J Chin Univ 30:92–95
Li ZF, Li HY, Liu YZ, Shi XN, Tang HA (2009) Chin Chem Bull 54:3014–3022
Alkorta I, Blanco F, Solimannejad M, Elguero J (2008) J Phys Chem A 112:10856–10863
Li Q, Xu X, Liu T, Jing B, Li W, Cheng J, Gong B, Sun JC (2010) Phys Chem Chem Phys 12:6837–6843
Li QZ, Jing B, Li R, Liu ZB, Li WZ, Luan F, Cheng JB, Gong BA, Sun JZ (2011) Phys Chem Chem Phys 13:2266–2271
Li QZ, Zhao JL, Jing B, Li R, Li WZ, Cheng JB (2011) J Comput Chem 32:2432–2440
Blanco F, Alkorta I, Solimannejad M, Elguero J (2009) J Phys Chem A 113:3237–3244
Zhao Q, Feng D, Sun Y, Hao J, Cai Z (2011) J Mol Model 17:1935–1939
Zabaradsti A, Kakanejadifard A, Ghasemian M (2012) Comput Theor Chem 989:1–6
Zhang ZF, Shen J, Jin NZ, Chen LP, Yang ZY (2012) Comput Theor Chem 999:48–54
Panek JJ, Berski S (2008) Chem Phys Lett 467:41–45
Roohi H, Nowroozi A, Eshghi F (2010) Int J Quantum Chem 110:1489–1499
Greenhill JV, Lue P (1993) Prog Med Chem 30:203–326, and references cited herein
Hollingworth RM (1976) Environ Health Persp 14:57–69
Lim JH, Lee EK, Kim Y (1997) J Phys Chem A 101:2233–2239
Kim Y, Lim S, Kim Y (1999) J Phys Chem A 103:6632–6637
Walewski L, Smaga A, Lesyng B, Sadlej J (2012) J Phys Chem A 116:10412–10419
Li P, Bu YX, Ai HQ, Yan SH, Han KL (2004) J Phys Chem B 108:16976–16982
Bell RL, Truong TN (1994) J Chem Phys 101:10442–10451
Siegbahn PEM, BlombergM RA, Crabtree RH (1997) Theor Chem Acc 97:289–300
Boys SF, Bernardi F (1970) Mol Phys 19:553–558
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian09, Revision A.02. Gaussian Inc, Wallingford
Reed AE, Curtiss LA, Weinhold F (1988) Chem Rev 88:899–926
Bader RFW (1990) Atoms in molecules: a quantum theory. Oxford University Press, Oxford, UK
Bulat FA, Toro-Labbé A, Brinck T, Murray JS, Politzer P (2010) J Mol Model 16:1679–1691
Murray JS, Politzer P (2013) ChemPhysChem 14:278–294
Politzer P, Murray JS, Clark T (2013) Phys Chem Chem Phys 15:11178–11189
Berski S, Silvi B, Latajka Z, Leszczynski J (1999) J Chem Phys 111:2542–2555
Politzer P, Riley KE, Bulat FA, Murray JS (2012) Comp Theor Chem 998:2–8
Koch U, Popelier PLA (1995) J Phys Chem A 99:9747–9754
Lipkowski P, Grabowski SJ, Robinson TL, Leszczynski J (2004) J Phys Chem A 108:10865–10872
Arnold WD, Oldfield E (2000) J Am Chem Soc 122:12835–12841
Espinosa E, Molins E, Lecomte C (1998) Chem Phys Lett 285:170–173
Acknowledgments
This work was supported by the National Natural Science Foundation of China (20973149), the Outstanding Youth Natural Science Foundation of Shandong Province (JQ201006), and the Program for New Century Excellent Talents in University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
An, X., Zhuo, H., Wang, Y. et al. Competition between hydrogen bonds and halogen bonds in complexes of formamidine and hypohalous acids. J Mol Model 19, 4529–4535 (2013). https://doi.org/10.1007/s00894-013-1969-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-013-1969-7