
Abstract
We presents a chemometrical study of the intermolecular properties of the C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes with X = F, CN, NC, and CCH. Through the MP2 perturbation theory and B3LYP hybrid functional, as well as modifications on 6-31ijGk basis sets with i = triple-zeta, j = diffuse and k = polarization functions, systematic tendencies in the R(n⋅⋅⋅HX) hydrogen bond distances and υ(n⋅⋅⋅HX) stretch frequencies were determined by the hierarchical cluster analysis, two level factorial designs and principal component analysis. Based on well-fitted math models, not only because polarization functions provide a great variance on statistical analysis, but this basis set reproduces more efficiently the available experimental results. Moreover, independent of whether the quality on basis set is increased, the effects yielded by both DFT and MP2 were not considered important in the statistical analysis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The viability to investigate properties of electronic structure with the purpose to interpret and justify their molecular phenomena is, traditionally, one cornerstone of chemistry. In computational chemistry, the correct choice of an adequate ab initio basis to study a molecular system still is considered an intriguing point among the theoreticians. Although some works reports that an appropriate basis set can describe traditional phenomena or else propose new insights with plausible perspectives [1–3], there is not a procedure to elect the ideal basis set, what in some cases can lead to an arbitrary choice of this parameter. However, some statistical studies have revealed the remarkable influence of the basis sets and quantumchemical methods on analysis of molecular properties of several systems [4–10], including those formed by hydrogen bonds [11, 12]. Nevertheless, it is well-known that in comparison with results obtained from HF (Hartree-Fock) [13] calculations, the molecular properties of hydrogen complexes [14] are described more efficiently by means of thesecond-order Møller-Plesset perturbation theory (MP2) [15] method. On the other hand, it has been documented that density functional theory (DFT) [16, 17] is also an efficient method, where its potentiality has been satisfactorily demonstrated in several promulgated studies, as for example in analysis of properties of hydrogen-bonded complexes [18–33]. Although it is well-established that both MP2 and DFT are the most popular methods used to evaluate the electronic correlation effects [34], it has been observed that DFT appears as an alternative computational scheme at the ab initio formalism [35] because the efficiency of exchange, correlation and hybrid functionals has been successfully demonstrated, in particular for studies of chemical, physical and biological systems [36–38].
In practice, Hohenberg-Kohn-Sham protocol offers a great advantage in comparison with the ab initio methods, one of them is the low computational demand. In studies of molecular properties of hydrogen-bonded complexes [39, 40], for instance, it is vital to consider the effects of electronic correlation, although some theoreticians are still not convinced that DFT is an able method due to its limitation for describing dispersion forces [41]. In spite of this, we are regarding a traditional question related at studies of hydrogen-bonded complexes: what is the most appropriated method for modeling these systems, MP2 or DFT? Or else, the efficiency of these two methods is influenced by any ab initio basis sets? So, by taking into account these two highlighted questions, this current study was elaborated with the proposal of comparing the efficiency of MP2 and DFT methods through the analysis of R(n⋅⋅⋅HX) hydrogen bond distances and υ(n⋅⋅⋅HX) infrared intermolecular frequencies of the C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes with X = F, CN, NC, and CCH (see Fig. 1). However, it is essential not only to evaluate which method is the most efficient, but we are admitting the importance of the basis sets and its applicability will be also discussed here. Concomitantly, it is necessary to use a combination of 6-31ijGk split-valence basis set with i = triple-zeta (Tξ); j = diffuse basis set (++), and k = polarization functions (d,p). Thereby, it is generated an immense quantity of results for R(n⋅⋅⋅HX) and υ(n⋅⋅⋅HX), which will be treated through the chemometric techniques, such as HCA (Hierarchical Cluster Analysis) [42], TLFD (Two Levels Factorial Design) [43, 44] and PCA (Principal Component Analysis) [45]. As it is well-known that chemometric techniques has been applied successfully in an immensity of situations where questions of chemical, physical and/or biological nature needs to be solved [46], it is necessary to mention that regarding to the main purpose of this work, we thinks that a chemometrical study of intermolecular properties of the C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes seems to be interesting by one single reason: due the ability of three-membered rings to form intermolecular complexes [47–50] and related systems [51, 52], an indication concerning of what level of theory can be appropriate to examine rings like C2H4O and C2H5N, in fact this is prerequisite to gain useful information about these systems.

Optimized geometries of the C2H4O⋅⋅⋅HF, C2H4O⋅⋅⋅HCN, C2H4O⋅⋅⋅HNC, C2H4O⋅⋅⋅HCCH, C2H5N⋅⋅⋅HF, C2H5N⋅⋅⋅HCN, C2H5N⋅⋅⋅HNC, and C2H5N⋅⋅⋅HCCH hydrogen-bonded complexes. In both R(n⋅⋅⋅HX) and υ(n⋅⋅⋅HX) terms, n symbolizes the lone electron pairs of the oxygen or nitrogen atoms in the C2H4O and C2H5N heterocyclics, respectively
Chemometric techniques
HCA
HCA is a statistical methodology that forms conglomerates patterns through the association among single samples. For the proposal of this work, the HCA similarity S ij of the theoretical methods and basis sets will be constructed on the basis of Euclidian criterion according to:
where d ab is the distance between a and b samples, as well as the dmax is the maximum distance for any connected point. Even though qualitatively, the application of HCA can give us a description of similarity among the theoretical levels used to study the R(n⋅⋅⋅HX) hydrogen bond distances and υ(n⋅⋅⋅HX) stretch frequencies of the C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes.
TLFD
To obtain a chemometrical model formed justly by relevant effects, the TLFD technique performs a complete variation in theoretical methods with the purpose to quantify the contributions of DFT and MP2, as well as of basis sets. Thus, we admitted the Pople’s 6-3ijGk [53] split-valence as our standard basis set and considering both DFT and MP2, the TLFD modifications are treated in four steps summarized below:
- (I) i :
-
Using triple-zeta (Tξ) instead of double (Dξ) valence functions [54];
- (II) j :
-
Including or not diffuse functions (++) [55];
- (III) k :
-
Adding or not polarization functions (d,p) [56];
- (IV) l :
-
Performing DFT instead of MP2 calculations.
Through these factors, it is built a simplified mathematical model that includes the most relevant effects Uk:
In Eq. (2) UEST and ŪX corresponds to estimated and medium values of the intermolecular properties R(n⋅⋅⋅HX) or υ(n⋅⋅⋅HX) of the C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes. Moreover, the k coefficient is a dummy variable, which during the analysis is equal to +1 and −1 when the corresponding factor (i, j, k or l) is present or absent, respectively.
PCA
PCA are vectors obtained from the diagonalization of X t X covariance matrix, where X are the original data represented in a multidimensional space [57, 58]. In the present case, the X matrix has 16 rows (theoretical levels, see TLFD section) and 4 columns (C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes with X = F, CN, NC or CCH, see Fig. 1), where the largest eigenvalue of X t X is interpreted as statistical information explained by the first principal component (Θ1), which if possible, describes the maximum variance (MV) governed by Eq. (3).
If the variance result is unsatisfactory, it is request a second principal component (Θ2), which is computed automatically. This procedure is repeated until to obtain the so desired maximum variance.
Computational scheme and statistical programs
To optimize the geometries of the C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes, all theoretical calculations related to the MP2 and DFT methods, as well as the 6-3ijGk basis sets were performed on the GAUSSIAN 98W [59] software. For DFT calculations, it was used the B3LYP hybrid [60], whose formalism is defined by the Becke’s three functional parameter [61] combined with the LYP non-local correlation term [62]. The results of the TLFD, HCA and PCA were obtained by the programs FATORIAL 1.0 [63, 64], STATISTICA 5.0 [65] and UNSCRAMBLER 7.5 [66], respectively.
Results
Structural parameter: R(n⋅⋅⋅HX) hydrogen bond distance
HCA
The Table 1 presents the theoretical values of the R(n⋅⋅⋅HX) hydrogen bond distances of the C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes. Initially, the application of HCA provided tendencies in R(n⋅⋅⋅HX) values, as can be observed in Fig. 2. Named as G2, this cluster indicates a great similarity among data obtained by the (d,p) polarization functions. However, G1 and G3 clusters are also formed and some interesting informations can be extracted from them. Note that, G1 corresponds basically to (++) diffuse functions, whereas G3 to smaller basis sets at MP2 level. If B3LYP is concerned, small basis sets without (++) diffuse functions leads to results similar to MP2, so that the results fall in G1 rather than G3. This is known and, if we compare with post-HF methods [67], it is well established that B3LYP was implemented with purpose to execute calculations by using moderate size basis sets instead of larger ones.

HCA graph of the R(n⋅⋅⋅HX) theoretical hydrogen bond distances of the C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes
TLFD
The TLFD results are listed in Table 2. We can observe that (d,p) polarizations and B3LYP functional are the most significant effects, whose contributions yields a systematic increases on R(n⋅⋅⋅HX) distances in 0.040 Å and 0.093 Å, as well as diminishes in −0.020 Å and -0.026 Å for the C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes, respectively. However, even though by increasing R(n⋅⋅⋅HX) in 0.024 Å and 0.025 Å, the interaction effect [(B3LYP)-(d,p)] provides a significant contribution to the TLFD analysis. Thus, the TLFD models for R(n⋅⋅⋅HX) hydrogen bond distances of the C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes are represented by the Eqs. (5) and (6), respectively.
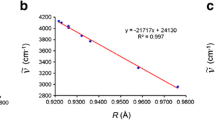
Note that, some other effects were also included in TLFD, such as (Tξ) valence and (++) diffuse functions. As can be seen in Fig. 3, the R2 linear correlation square coefficient of 0.99 indicates a good agreement between the \({\text{R}}_{\left( {{\text{n}} \cdots {\text{HX}}} \right)}^{{\text{EST}}} \) estimated results and \(\overline{{\text{R}}} _{{_{{{\text{X}}{\kern 1pt} _{{{\left( {{\text{n}}...{\kern 1pt} {\text{HX}}} \right)}}} }} }} \) medium values of the hydrogen bond distances of the C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes.

Plot of the \(R_{\left( {n \cdots HX} \right)}^{{\text{EST}}} \) estimated hydrogen bond distances versus the correspondent R(n⋅⋅⋅HX) theoretical values of the C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes
PCA
In terms of PCA, according to Figs. 4 and 5, it can be seen that (d,p) polarizations are described by Θ1 axis, which accounts for 80% and 81% of variance for the R(n⋅⋅⋅HX) hydrogen bond distance values of the correspondent C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes. It should be also noticeable that, as Θ2 explains 14% and 16% of variance, we would like to emphasize that meaningful effects are also explained by Θ2, for instance the (++) diffuse functions. Indeed, the PCA analysis yielded a satisfactory description of variance if the results of second component are included into the chemometrical analysis. As such, both Θ1 and Θ2 account for 94% and 97% of the total variance.

PCA scores of the R(n⋅⋅⋅HX) theoretical hydrogen bond distances of the C2H4O⋅⋅⋅HX hydrogen-bonded complexes

PCA scores of the R(n⋅⋅⋅HX) theoretical hydrogen bond distances of the C2H5N⋅⋅⋅HX hydrogen-bonded complexes
Not only in terms of (d,p) polarization functions, which appropriately describes the polarizability and strain energies of the C2H4O and C2H5N heterorings, but the great importance of (++) diffuse functions is related with its capacity of describing chemical systems with larger spatial region and trends of expansion due to their electrons are placed relatively far from the nuclei, i.e., molecules with electron pairs or even anions [68]. For the analysis of the R(n⋅⋅⋅HX) values, however, these concepts makes reference to n lone electron pairs of oxygen and nitrogen atoms on C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes, respectively. In regards to the acids, the loading Eqs. (7), (8), (9), and (10) demonstrates that hydrofluoric acid is the most important proton donor due to its great contribution for the variance data in both Θ1 and Θ2 axis.
However, the validation of the chemometrical model still requires a comparative study with experimental data. Some time ago, the experimental geometries of the C2H4O⋅⋅⋅HF and C2H4O⋅⋅⋅C2H2 hydrogen-bonded complexes were explained by Legon et al. [69, 70] through Fourier transform microwave spectroscopy (FTMS) [71] analysis. In these occasions, the values of 1.70 Å and 2.40 Å for the R(n⋅⋅⋅HX) hydrogen bond distances of the C2H4O⋅⋅⋅HF and C2H4O⋅⋅⋅C2H2 complexes were determined. By analyzing the values listed in Table 2, it is clearly perceived that (d,p) polarizations provide the best concordance of R(n⋅⋅⋅HX) theoretical hydrogen bond distances in comparison with the FTMS values. For instance, if we make a comparison between MP2/6-31G(d,p) and B3LYP/6-31G(d,p) calculations, which yielded results of 1.7112 Å and 1.7010 Å for C2H4O⋅⋅⋅HF, as well as 2.1363 Å and 2.1275 Å for the C2H4O⋅⋅⋅ C2H2 complexes, it must be noted that these theoretical results are in good agreement with the experimental data of 1.70 Å and 2.40 Å presented above. In this sense, it can be noted that MP2 and B3LYP are considered methods of comparable accuracy. As these are the available experimental data [69, 70], it is with the understanding that such direct comparison is the only test of theory.
Vibrational harmonic spectrum: υ(n⋅⋅⋅HX) hydrogen bond stretch frequencies
HCA
Table 3 presents the theoretical values of the υ(n⋅⋅⋅HX) hydrogen bond stretch frequencies of the C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes. Likewise to structural analysis, the same procedure was employed to examine the theoretical values of the hydrogen bonds frequencies, i.e, the HCA, FDTL and PCA techniques were applied in sequence. For HCA evaluation, the tree cluster presented in Fig. 6 characterizes a G1 group basically formed by (d,p) polarization functions. For remaining factors, neither similarity was observed as very important.

HCA graph of the theoretical υ(n⋅⋅⋅HX) stretch frequencies of the C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes
TLFD
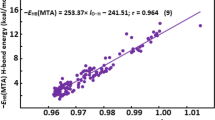
Nevertheless, Table 4 lists the TLFD results of the υ(n⋅⋅⋅HX) hydrogen bonds frequencies of the C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes. As we can see, the main effects are indicated by (d,p) polarizations and [(B3LYP)-(d,p)] interaction effects, which decreases systematically υ(n⋅⋅⋅HX) in −8.9 cm−1 and −29.3 cm−1, as well as in −5.4 cm−1 and −7.2 cm−1 for the C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes, respectively. Rather, these effects and others were used to build two TLFD models represented by Eqs. (11) and (12), where the R2 linear correlation square coefficient of 0.98 indicates a good concordance between \(\upsilon _{\left( {n \cdots HX} \right)}^{{\text{EST}}} \) and \(\bar \upsilon {\kern 1pt} _{{\text{X}}_{\left( {{\text{n}} \cdots {\text{HX}}} \right)} }^{} \) values of the C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes, as can be seen in Fig. 7.

Plot of the \(\upsilon _{\left( {n \cdots HX} \right)}^{{\text{EST}}} \) estimated hydrogen bond stretch frequencies versus the correspondent υ(n⋅⋅⋅HX) theoretical values of the C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes
PCA
About PCA, however, the results shows that (d,p) polarization functions and [(B3LYP)-(d,p)] interaction are the most prominent effects, which describes 72% and 80% of variance through the Θ1 axis, as can be seen in Figs. 8 and 9, respectively. Both (d,p) and [(B3LYP)-(d,p)] are placed in left side of Θ1 and Θ2, although the second component contains 24% and 19% of variance for υ(n⋅⋅⋅HX) values of the C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes, respectively. Corroborating with the structural analysis, loading Eqs. (13), (14), (15), and (16) says that the hydrofluoric acid has the largest contribution on the variance data.
On the contrary of results found for the hydrogen bond distances, the (d,p) polarizations are identified in the left side of both Θ1 and Θ2 axis. In other words, our model can predict that weaker hydrogen bond frequencies are obtained through the application of (d,p) polarization functions. Comparatively, it was verified that MP2 contributes slightly to the statistical model. It is, in fact, not a surprise because DFT simulates molecular parameters of cyclic hydrogen-bonded complexes very satisfactorily if compared with MP2 [72], or else still more efficiently than HF calculations [73].

PCA scores of the υ(n⋅⋅⋅HX) theoretical stretch frequencies of the C2H4O⋅⋅⋅HX hydrogen-bonded complexes

PCA scores of the υ(n⋅⋅⋅HX) theoretical stretch frequencies of the C2H5N⋅⋅⋅HX hydrogen-bonded complexes
Discussion and perspectives
It is a tradition in our research group to use DFT calculations in studies of hydrogen-bonded complexes [74, 75] and related systems [76]. As reviewed on a number of occasions, it is widely known the efficiency of the B3LYP hybrid functional for describing intermolecular interactions [77–80] but independent of that, it is well to bear in mind that MP2 is considered an unquestionable approach, wherein it was also used in some investigations developed by our research group [14, 81]. So, we have no desire to opine or to enter a discussion if DFT is better than MP2 and vice-versa, actually we just want to discern the efficiency of these two methods for describing the intermolecular properties of the C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes. Some time ago, we published some works wherein structural, electronic, and vibrational parameters of these complexes were debated. In these works, of course that the B3LYP was admitted but, it used the 6-311 +G(d,p) basis sets to effectuate the calculations, by which is a complete set because there is valence (Tξ) and diffuse (++) functions within its formulation. If we considered the results of our chemometrical analysis, by which it was concluded that (d,p) functions is the most important parameter, it was redundant to include (Tξ) and (++) into our works cited above? [72, 82]. In parts, actually, but on the other hand there is the problem of base sets superposition error (BSSE) [83], wherein if small basis are used, such as 6-31G(d,p), surely that BSSE amounts will be overestimated [84], e.g., the dispersion effects caused by moderate basis sets leads to a correction error on the interaction energy of 8.26–1.55 kJ mol−1, as recently reported by Šponer and Hobza [85]. It is by this statement that, at this moment we are developing a study wherein it is evaluated the dependence of BSSE with the same ab initio basis sets used in this work, properly not just related to the C2H4O⋅⋅⋅HX or C2H5N⋅⋅⋅HX systems but, we are extending this study to other hydrogen-bonded complexes with significant differences of intermolecular strength, such as those formed by π and pseudo-π interactions [25, 83–94]. So, as soon as possible, we expect to obtain a measure of how the basis sets can contribute for BSSE calculations. It comes to what is the most important is that the relationship between BSSE and basis sets is well understood but, in a similar way at procedure performed here where DFT and MP2 methods were used, it is worth stressing that this perspective above presented can reveal interesting results, although there are some affirmations about a possible relationship between both DFT and MP2 with accurate BSSE results [95, 96].
Conclusions
The HCA, TLFD and PCA chemometric techniques were used to evaluate the effects of basis sets and quantumchemical methods on structural parameters and infrared harmonic spectrum of C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX hydrogen-bonded complexes. These three statistical methods were applied with the purpose to ensure that an appropriate level of theory can be elected to describe a property of interest, in such case, the intermolecular distance and its stretch frequency of the complexes mentioned above. In general, the results of our study show that the polarization functions are the most important effect, although other factors were also considered significant, for instance diffuse functions on the analysis of the hydrogen bonds distance. At the interpretation of the hydrogen bonds frequencies, polarization functions and B3LYP functional contributes equally for the statistical analysis upon removal of the other effects. In comparison with HCN, HNC, and HCCH, the PCA results demonstrated that the hydrofluoric acid is the most important variable. Finally, a decisive point is related to the quality of theoretical method, DFT or MP2. In this sense, it was verified that MP2 results alters insignificantly the statistical analysis. Finally we comment the relation of our current results with theoretical works documented recently [11, 12], where it was known that MP2 always furnished the major contribution to the chemometrical analysis. By taking into account our results debated in this work, DFT seems to be a suitable and efficient approach to study intermolecular properties of hydrogen-bonded complexes [97–100]. Thereby, we would like to emphasize the importance of DFT as a useful method for studying electronic structures and in this sense, only two reasons deserve to be considered: i) the lower computational effort and, ii) satisfactory reproduction of the available experimental data.
References
Sordo TL, Barrientos C, Sordo JA, Fraga S (1992) Computational chemistry: structure, interactions and reactivity Part A. Elsevier, Amsterdam
Sordo TL, Sordo JA (1992) J Mol Struct THEOCHEM 253:17–23. doi:10.1016/0166-1280(92)87094-G
Helgaker T, Taylor PR, Yarkoni DR (1995) Modern electronic structure theory Part II. Word Scientific, Singapore
Suto E, Ferreira MMC, Bruns RE (1991) J Comput Chem 12:885–890. doi:10.1002/jcc.540120715
Azevedo ALMS, Neto BB, Scarminio IS, Oliveira AE, Bruns RE (1995) J Comput Chem 17:167–177. doi:10.1002/(SICI)1096-987X(19960130)17:2<167::AID-JCC4>3.0.CO;2-U
da Silva JBP, Ramos MN, Bruns RE (1997) Spectrochim Acta [A] 53:733–747. doi:10.1016/S1386-1425(96)01790-8
Ramos MN, da Silva JBP, Bruns RE (▪▪▪) Spectrochim Acta [A] 53:1563–1579. doi:10.1016/S1386-1425(97)00031-0
da Silva JBP, Neto BB, Ramos MN, Bruns RE (1998) Chemom Intell Lab Syst 44:187–195. doi:10.1016/S0169-7439(98)00121-X
Ramos MN, Lopes KC, Monte SA, Ventura E, Araújo RCMU (2006) J Mol Struct THEOCHEM 760:21–27. doi:10.1016/j.theochem.2005.10.029
Silva JBP, Ramos MN (2001) J Mol Struct THEOCHEM 539:83–92. doi:10.1016/S0166-1280(00)00775-2
Araújo RCMU, da Silva JBP, Neto BB, Ramos MN (2002) Chemom Intell Lab Syst 62:37–46. doi:10.1016/S0169-7439(01)00213-1
Ramos MN, Lopes KC, Pereira FS, Araújo RCMU, Bruns RE (2004) Chemom Intell Lab Syst 70:157–163. doi:10.1016/j.chemolab.2003.11.004
Roothaan CCJ (1951) Rev Mod Phys 23:69–89. doi:10.1103/RevModPhys.23.69
Araújo RCMU, da Silva JBP, Ramos MN (1995) Spectrochim Acta [A] 51:821–830. doi:10.1016/0584-8539(94)00194-G
MØller C, Plesset M (1934) Phys Rev 56:618–622
Hohenberg P, Kohn W (1964) Phys Rev B 136:864–871. doi:10.1103/PhysRev.136.B864
Kohn W, Sham LJ (1965) Phys Rev A 140:1133–1138. doi:10.1103/PhysRev.140.A1133
Oliveira BG, Vasconcellos MLAA (2006) J Mol Struct THEOCHEM 774:83–88. doi:10.1016/j.theochem.2006.06.018
Oliveira BG, Araújo RCMU, Carvalho AB, Lima EF, Silva WLV, Ramos MN, Tavares AM (2006) J Mol Struct THEOCHEM 775:39–45. doi:10.1016/j.theochem.2006.06.028
Oliveira BG, Araújo RCMU, Carvalho AB, Ramos MN (2007) J Theor Comput Chem 6:647–660. doi:10.1142/S0219633607003362
Oliveira BG, Araújo RCMU, Carvalho AB, Ramos MN (2007) Chem Phys Lett 433:390–394. doi:10.1016/j.cplett.2006.11.029
Oliveira BG, Pereira FS, Araújo RCMU, Ramos MN (2006) Chem Phys Lett 427:181–184. doi:10.1016/j.cplett.2006.06.019
Oliveira BG, Araújo RCMU, Carvalho AB, Ramos MN (2007) Spectrochim Acta [A] 68:626–631. doi:10.1016/j.saa.2006.12.038
Zheng Y-J, Bruice TC (1997) Proc Natl Acad Sci USA 94:4285–4288. doi:10.1073/pnas.94.9.4285
Chandra AK, Nguyen MT (1998) Chem Phys 232:299–306. doi:10.1016/S0301-0104(98)00111-6
Kryachko ES, Nguyen MT (2001) J Chem Phys 115:833–841. doi:10.1063/1.1371516
Lamarche O, Platts JA (2002) Chem Eur J 8:457–466. doi:10.1002/1521-3765(20020118)8:2<457::AID-CHEM457>3.0.CO;2-5
Ireta J, Neugebauer J, Scheffler M (2004) J Phys Chem A 108:5692–5698. doi:10.1021/jp0377073
Vila A, Graña AM, Mosquera RA (2002) Chem Phys 281:11–22. doi:10.1016/S0301-0104(02)00590-6
van Erp TS, Meijer EJ (2003) J Chem Phys 118:8831–8840. doi:10.1063/1.1567258
Qu Z-W, Zhu H, Zhang X-K, Zhang Q-Y (2003) Chem Phys Lett 367:245–251. doi:10.1016/S0009-2614(02)01708-6
Zhao Y, Tishchenko O, Truhlar DG (2005) J Phys Chem B 109:19046–19051. doi:10.1021/jp0534434
Alonso JA, Mañanes A (2007) Theor Chem Acc 117:467–472. doi:10.1007/s00214-006-0079-3
Lowdin PO (1959) Adv Chem Phys 2:207–322. doi:10.1002/9780470143483.ch7
Hammes-Schiffer S, Andersen HC (1993) J Chem Phys 99:523–532. doi:10.1063/1.465776
Hedin L, Lundvist B (1971) J Phys C Solid State Phys 4:2064–2083. doi:10.1088/0022-3719/4/14/022
Geerlings P, De Proft F, Langenaeker W (2003) Chem Rev 103:1793–1874. doi:10.1021/cr990029p
Perdew JP, Yue W (1986) Phys Rev B 33:8800–8802. doi:10.1103/PhysRevB.33.8800
Belosludov RV, Li Z-Q, Kawazoe Y (2004) Mol Eng 8:105–120. doi:10.1023/A:1008334322264
Park YC, Lee JS (2007) Bull Korean Chem Soc 28:386–390
Holroyd LF, van Mourik T (2007) Chem Phys Lett 442:42–46. doi:10.1016/j.cplett.2007.05.072
Almeida JAS, Barbosa LMS, Pais AACC, Formosinho SJ (2007) Chemom Intell Lab Syst 87:208–217. doi:10.1016/j.chemolab.2007.01.005
Liao C-T (2006) J Stat Plann Inf 136:4071–4087. doi:10.1016/j.jspi.2005.04.005
Montgomery CD (1996) Design and analysis of experiments. Wiley, New York
Mackiewicz A, Ratajczak W (1993) Comput Geosci 19:303–342. doi:10.1016/0098-3004(93)90090-R
Matter H, Schwab W (1999) J Med Chem 42:4506–4523. doi:10.1021/jm990250u
Delanoye SN, Herrebout WA, van der Veken BJ (2005) J Phys Chem A 109:9836–9843. doi:10.1021/jp051476d
Herrebout WA, Delanoye SN, Maes BUW, van der Veken BJ (2006) J Phys Chem A 110:13759–13768. doi:10.1021/jp065502z
Craw JS (1991) J Chem Soc, Faraday Trans 87:3495–3497. doi:10.1039/ft9918703495
Borho N, Xu Y (2007) Phys Chem Chem Phys 9:4514–4520. doi:10.1039/b705746f
Piancastelli MN, Bao Z, Hennies F, Travnikova O, Céolin D, Kampen T, Horn K (2007) Appl Surf Sci 254:108–112. doi:10.1016/j.apsusc.2007.07.070
Ferraz AC, Miotto R (2007) Surf Sci 601:2576–2579. doi:10.1016/j.susc.2006.11.081
Ditchfield R, Hehre WJ, Pople JA (1971) J Chem Phys 54:724–728. doi:10.1063/1.1674902
Krishnan R, Frisch MJ, Pople JA (1980) J Chem Phys 72:4244–4245. doi:10.1063/1.439657
Clark T, Chandrasekhar J, Spitznagel GW, Schleyer Pv R (1983) J Comput Chem 4:294–301. doi:10.1002/jcc.540040303
Frisch MJ, Pople JA, Binkley JS (1984) J Chem Phys 80:3265–3269. doi:10.1063/1.447079
Teppola P, Mujunen SP, Minkkinen P, Puijola T, Pursiheimo P (1998) Chemom Intell Lab Syst 44:307–317. doi:10.1016/S0169-7439(98)00188-9
Jolliffe IT (2002) Principal component analysis. Springer, New York
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA Jr, Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Rega N, Salvador P, Dannenberg JJ, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Baboul AG, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andres JL, Gonzalez C, Head-Gordon M, Replogle ES, Pople JA (1998) GAUSSIAN 98W (Revision A.1). Gaussian, Inc., Pittsburgh PA
Becke AD (1997) J Chem Phys 107:8554–8560. doi:10.1063/1.475007
Becke AD (1993) J Chem Phys 98:5648–5652. doi:10.1063/1.464913
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785–789. doi:10.1103/PhysRevB.37.785
Scarmínio IS, Bruns RE (1989) Trends Anal Chem 8:326–327. doi:10.1016/0165-9936(89)87038-4
Neto BB, Scarmínio IS, Bruns RE (1995) Planejamento e Otimização de Experimentos, Ed. Unicamp, Campinas, Brazil
Statistica for Windows (1995) StatSoft, Inc., 2325 East 13th Street, 74104, Tulsa, OK
The Unscramlber v 7.5 (1999) CAMO ASA, Box 1405 Vika, N-0115 Oslo, Norway
Simons J, Jordan KD (1987) Chem Rev 87:535–555. doi:10.1021/cr00079a004
Leyssens T, Peeters D (2004) J Mol Struct THEOCHEM 673:79–86. doi:10.1016/j.theochem.2003.12.001
Legon AC, Wallwork AL, Milen DJ (1991) Chem Phys Lett 178:279–284. doi:10.1016/0009-2614(91)87069-N
Legon AC (1995) Chem Phys Lett 247:24–31. doi:10.1016/0009-2614(95)01172-9
Legon AC (1983) Annu Rev Phys Chem 34:275–300. doi:10.1146/annurev.pc.34.100183.001423
Oliveira BG, Santos ECS, Duarte EM, Araújo RCMU, Ramos MN, Carvalho AB (2004) Spectrochim Acta [A] 60:1883–1887. doi:10.1016/j.saa.2003.10.006
Rozas I, Alkorta I, Elguero J (1997) J Phys Chem A 101:9457–9463. doi:10.1021/jp971893t
Oliveira BG, Araújo RCMU, Ramos MN (2008) Struct Chem 19:185–189. doi:10.1007/s11224-007-9269-4
Oliveira BG, Araújo RCMU, Ramos MN (2008) Struct Chem 19:665–670. doi:10.1007/s11224-008-9344-5
Filho EBA, Ventura E, do Monte SA, Oliveira BG, Junior CGL, Rocha GB, Vasconcellos MLAA (2007) Chem Phys Lett 449:336–340
Oliveira BG, Araújo RCMU (2007) Quim Nova 30:791–796
Oliveira BG, Araújo RCMU, Ramos MN (2007) Quim Nova 30:1167–1170
Oliveira BG, Araújo RCMU, Pereira SF, Lima EF, Silva WLV, Carvalho AB, Ramos MN (2008) Quim Nova 31:1673–1679
Oliveira BG, Araújo RCMU, Chagas FF, Carvalho AB, Ramos MN (2008) J Mol Model 14:949–955. doi:10.1007/s00894-008-0337-5
Araújo RCMU, Ramos MN (1996) J Mol Struct THEOCHEM 366:233–240. doi:10.1016/0166-1280(96)04536-8
Oliveira BG, Duarte EM, Araújo RCMU, Ramos MN, Carvalho AB (2005) Spectrochim Acta [A] 61:491–494. doi:10.1016/j.saa.2004.04.023
Boys SB, Bernardi F (1970) Mol Phys 19:553–566. doi:10.1080/00268977000101561
Kobko N, Dannenberg JJ (2001) J Phys Chem A 105:1944–1950. doi:10.1021/jp001970b
Šponer J, Hobza P (2000) J Phys Chem A 104:4592–4597. doi:10.1021/jp9943880
Chandra AK, Pal S, Limaye AC, Gadre SR (1995) Chem Phys Lett 247:95–100. doi:10.1016/0009-2614(95)01175-4
Tang T-H, Cui Y-P (1996) Can J Chem 74:1162–1170. doi:10.1139/v96-130
Zhang Y-H, Hao J-K, Wang X, Zhou W, Tang T-H (1998) J Mol Struct THEOCHEM 455:85–99. doi:10.1016/S0166-1280(98)00247-4
Wojtulewski S, Grabowski S (2002) J Mol Struct 605:235–240. doi:10.1016/S0022-2860(01)00785-2
Grabowski SJ (2004) J Phys Chem A 108:1806–1812. doi:10.1021/jp036770p
Grabowski SJ (2007) Chem Phys Lett 436:63–67. doi:10.1016/j.cplett.2007.01.041
Grabowski SJ (2007) J Phys Chem A 111:13537–13543. doi:10.1021/jp076990t
Grabowski SJ (2007) J Phys Chem A 111:3387–3393. doi:10.1021/jp070530i
Grabowski SJ, Sokalski WA, Leszczynski J (2006) Chem Phys Lett 432:33–39. doi:10.1016/j.cplett.2006.10.069
Daza MC, Dobado JA, Molina JM, Villaveces JL (2000) Phys Chem Chem Phys 2:4089–4094. doi:10.1039/b001885f
Shields AE, van Mourik T (2007) J Phys Chem A 111:13272–13277. doi:10.1021/jp076496p
Jalbout AF, Hameed AJ, Trzaskowski B (2007) J Mol Struct THEOCHEM 816:1–3. doi:10.1016/j.theochem.2007.03.037
Basch H, Hoz S (1997) J Phys Chem A 101:4416–4431. doi:10.1021/jp970011n
Chandra AK, Nguyen MT, Zeegers-Huyskens T (2000) J Mol Struct 519:1–11. doi:10.1016/S0022-2860(99)00276-8
Chandra AK, Michalska D, Wysokiñsky R, Zeegers-Huyskens T (2004) J Phys Chem A 108:9593–9600. doi:10.1021/jp040206c
Acknowledgements
The authors gratefully acknowledge partial financial support from Brazilian Funding agencies CAPES and CNPq.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Oliveira, B.G., Araújo, R.C.M.U., Carvalho, A.B. et al. A chemometrical study of intermolecular properties of hydrogen-bonded complexes formed by C2H4O⋅⋅⋅HX and C2H5N⋅⋅⋅HX with X = F, CN, NC, and CCH. J Mol Model 15, 421–432 (2009). https://doi.org/10.1007/s00894-008-0422-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-008-0422-9