
Abstract
High throughput docking (HTD) is routinely used for in silico screening of compound libraries with the aim to find novel leads in a drug discovery program. In the absence of an experimentally determined structure, a homology model can be used instead. Here we present an assessment of the utility of homology models in HTD by docking 300,000 anticipated inactive compounds along with 642 known actives into the binding site of the insulin-like growth factor 1 receptor (IGF-1R) kinase constructed by homology modeling. Twenty-one different templates were selected and the enrichment curves obtained by the homology models were compared to those obtained by three IGF-1R crystal structures. The results show a wide range of enrichments from random to as good as two of the three IGF-1R crystal structures. Nevertheless, if we consider the enrichment obtained at 2% of the database screened as a performance criterion, the best crystal structure outperforms the best homology model. Surprisingly, the sequence identity of the template to the target is not a good descriptor to predict the enrichment obtained by a homology model. The three homology models that yield the worst enrichment have the smallest binding-site volume. Based on our results, we propose ensemble docking to perform HTD with homology models.

Top-scoring binding mode of NVP-AEW541 found by Glide with the aew receptor
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The genome sequencing projects result in an ever increasing number of protein sequences. Structural genomics is expected to elucidate many experimentally determined protein structures and, therefore, representative structures for most of the protein families will soon be available. Nevertheless, it is unlikely that there will ever be an atomic resolution structure of every relevant protein. Furthermore, solving a protein structure by crystallography or NMR remains a long and expensive effort. Virtual screening by docking requires a three-dimensional (3D) structure of the target of interest. In this respect, constructing 3D-models based on the structures of homologous proteins represents an interesting approach.
Homology modeling has the potential to significantly impact the drug development cycle. (see [1, 2] for a review). One of the possible applications is the high throughput docking (HTD) of large databases of compounds. Several successful applications of HTD using comparative models have been published. Rhodopsin-based homology models of G-protein coupled receptors have been shown to be useful to enrich known active compounds in database screening [3] and to discover novel and potent ligands [4–6]. A potent and selective inhibitor of the protein casein kinase 2 was identified by HTD using a homology model [7]. An evaluation of the efficacy of docking to an homology model and to the crystal structure for 10 proteins was reported [8]. Although the best results were obtained with the crystal structure, the homology models yielded significant enrichment of known active compounds against a background of decoys. A HTD experiment of a database of 32,000 compounds with homology models was reported for six different kinases [9]. In five of the six cases, the known active compounds were found to be enriched by a factor of 5 in the top 5% of the database screened. A wide range of performance was also observed. Some homology models performed comparably to the crystal structures, whereas others performed significantly worse. In one case, the homology model even yielded better enrichment than the crystal structure [9]. The relationship between sequence identity and docking performance was addressed by carrying out HTD with eight homology models for factor VIIa and four homology models for cyclin-dependent kinase 2 (CDK2) [10]. For factor VIIa, homology models based upon templates with a sequence identity above 50% outperformed those built with templates with a sequence identity below 50%. For cyclin-dependent kinase 2, no correlation between sequence identity and enrichment factor was observed. Gilson et al. reported an in-depth assessment of the performance of HTD with comparative models for five targets [11]. Some homology models were found to yield an enrichment comparable to that obtained by the crystal structure. Furthermore, the sequence identity of the template used to build the homology to the target was found to show little correlation with the performance of the docking calculations. Very recently, a discovery of novel inhibitors of 12-and 15-human lipoxygenase by HTD with homology models was reported [12].
Here, we present an evaluation of the utility of homology models in HTD using the insulin-like growth factor 1 receptor kinase (IGF-1R) as the target protein. Although we investigated only one kinase, the results of this study are expected to be similar across the kinase family, given the high similarity of the ATP binding site. We selected the IGF-1R because kinases represent a very important class of drug targets. They are implicated in many diseases including cancer [13], diabetes [14], and inflammation [15]. Currently, around 20 percent of all research programs at major pharmaceutical companies are focused on kinases and they represent an important target for structure-based drug design (SBDD) [16–19]. Furthermore, the human genome contains a large number of protein kinases: 518 distinct human kinase genes have been identified [20]. As of June 2003, the Protein Data Bank (PDB) [21] contained 164 crystal structures of protein kinases, and only 38 of the 518 putative kinase targets were represented [22]. Therefore, homology modeling could significantly contribute to the discovery of novel kinase inhibitors. From a structural point of view, protein kinases share a common fold, which consists of two domains, an N-terminal domain and C-terminal domain connected via a so-called hinge-domain, which allows rotation of the two domains. The hinge domain is also involved in the binding of ATP via two hydrogen bonds. The conserved ATP binding site makes the design of selective inhibitors very challenging. On the other hand, it simplifies the alignment procedure involved in comparative modeling.
In this work, 21 templates were selected for homology model building, which guarantees statistically meaningful conclusions. A large number of compounds (300,000) from the Novartis Corporate Database, along with 642 known IGF-1R active compounds, were docked into the ATP binding site of the IGF-1R structures and the 21 homology models using the program Glide [23]. A sequence and structure analysis of the enrichment results is presented.
Methods
Target protein and homology modeling
The IGF-1R kinase belongs to the tyrosine protein kinase subfamily. Three IGF1-1R crystal structures were available at the time this study was undertaken. Two structures are publicly available: 1jqh, which was co-crystallized with ANP, an ATP analog, and 1p4o, which is an apo, inactive structure. The third structure is an in-house structure and was co-crystallized with the inhibitor NVP-AEW541, which binds IGF-1R with an IC50 of 0.15 μm in an in vitro assay [24] (Fig. 1). This structure will be referred to hereafter as aew. To select the templates, a basic local alignment search tool (BLAST) search against the PDB was conducted using the IGF-1R kinase sequence as a query. Only the structures with a Blast Bit-score above 130 were kept. The following criteria were then used to discard structures: (a) no ligand bound (apart from one structure, see below), (b) missing residues in the binding site, (c) inactive conformation, and (d) structural similarity of the binding site (for the structures sharing a very similar structural binding site, only one was kept to limit the number of homology models). The final selection of templates contains 21 structures, which all belong to the tyrosine protein kinase subfamily (Table 1). Among these templates, it is worth mentioning the insulin receptor (InsR) kinase, because it is very closely related to the IGF-1R. Only one residue (Thr in IGF-1R, which is mutated into Ala in InsR) that are in close proximity to ATP differs. Furthermore, this residue is located in the hinge and does not have a direct interaction with ATP. Two crystal structures of InsR are available in the PDB: 1ir3, which was co-crystallized with an ATP analog, and 1irk, which represents the unphosphorylated, apo form of InsR. Furthermore, Table 1 also shows that four templates were built upon the fibroblast growth factor receptor (FGFR) kinase co-crystallized with four different ligands.

Structure of NVP-AEW541
The homology models were built using Prime [25] accessible through the Maestro interface [26]. All water molecules were removed and the bound ligand was kept. The alignment of the binding site residues was always unambiguous, since there was no gap in the sequences for these residues. During the homology model building, Prime keeps the backbone rigid for the cases in which the backbone does not need to be reconstructed due to gaps in the alignment. Several measures of the similarity of the templates to the target are provided in Table 1: kinase domain and binding site sequence identity, number of gaps and insertions, and heavy atom root-mean-square deviation (RMSD) from the aew structure of the binding site residues. A residue was considered as a binding site residue if it had at least one heavy atom within 5 Å from the ligand bound in aew (NVP-AEW541). Apart from the InsR kinase, Table 1 shows that the kinase domain and binding site sequence identity range between 32% and 43%, and between 55% and 72%, respectively.
Known binders and database for high throughput docking
300,000 compounds were randomly selected from the Novartis Corporate Database and expressed as sdf file. For each compound, 3D coordinates were generated using CORINA [27]. The compound structures were then ionized assuming a pH of 7.0 with the ionizer module supplied by Schrödinger [28]. Compounds were finally subject to a minimization based on the OPLS-AA force-field. The 642 compounds that bind IGF-1R with an IC50 below 10 μm were also extracted from the Novartis Corporate Database. These compounds were grouped with the clustering method implemented in Pipeline Pilot [29], which is a maximal dissimilarity-based partitioning method. The maximum distance of any cluster member from the center of this cluster was set to 0.6 and the FCFP_6 descriptors were used. 85 clusters were obtained in this way, 32 of which contain only one member (singleton). This result shows the significant diversity of the IGF-1R actives.
Docking using Glide
All protein structures were prepared according to the standard procedure described in the Glide user manual [23]. The default input parameters implemented in Maestro were used for the generation of the command files for the docking of the small-molecule database against the IGF-1R crystal structures and homology models. For the generation of the scoring grid, the van der Waals radii of the non-polar protein atoms were not scaled. Compounds were docked using Glide in single-precision mode. In order to limit the number of false positive compounds, a pharmacophoric constraint (hydrogen bond with the NH of Met83, since all potent kinase inhibitors form this hydrogen bond) was used in the docking experiment. The best pose for each compound, as evaluated by the Glide scoring function, was written out.
Post-processing of HTD data
After the HTD experiment, the successfully docked compounds were sorted into a ranked list based upon the Glide scores. This ranked list was then used to generate the enrichment and receiver operating characteristic (ROC) curves [30]. These two curves are not equivalent. A ROC curve describes the tradeoff between sensitivity and specificity. Sensitivity is defined as the ability of the model to detect true positives while specificity is its ability to avoid false negatives. The area below a ROC curve can be used to quantify the enrichment. A ROC value greater than 0.9 is considered excellent and a value below 0.6 represents no enrichment.
Results and discussion
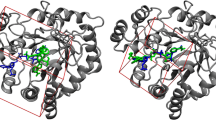
We first evaluated the ability of the three IGF-1R crystal structures to discriminate active compounds from inactive ones. Figure 2 shows the enrichment curves obtained by the Glide scoring function for these three receptors. They perform significantly better to pick up the active compounds than a random model. 1jqh and 1p4o yield similar enrichments and significantly worse than those that obtained by aew in the top 5% of the database. The better enrichment obtained by the aew structure originates from an induced-fit effect of NVP-AEW541 upon binding. Figure 3 shows the binding mode found by Glide with the aew and 1jqh receptors. For the aew receptor, the RMSD of the top-scoring pose from the crystal structure is only 0.7 Å and the Glide score is −10.4, whereas the corresponding values for the 1jqh receptor are 10.1 Å and −5.4, respectively. For the latter receptor, the aspartic acid that forms a salt bridge with the catalytic lysine would clash with the benzyloxy-phenyl group of NVP-AEW541 in the right binding mode, and Glide cannot generate this mode since the receptor is kept rigid upon docking. The benzyloxy-phenyl pyrrolo-pyrimidine moiety of NVP-AEW541 is present in 83 out of the 642 binders. For most of these compounds, Glide generates the correct binding mode with the aew receptor, whereas a wrong docked pose is obtained with the 1jqh and 1p4o receptors.

Enrichment curves for the three IGF-1R crystal structures. The curve “Perfect” is based on the assumption that all the compounds that have not been experimentally tested are inactive

Top-scoring binding mode of NVP-AEW541 found by Glide with the aew receptor (left) and with the 1jqh receptor (right)
Next, we analyze the ability of the homology models to retrieve the active compounds. The enrichment curves obtained by the models built upon the two InsR kinases and the four FGFR kinases are depicted in Fig. 4 and in Fig. 5, respectively. In both cases, the performance of the homology modes ranges from random to similar to that obtained by the 1jqh and 1p4o receptors. This result clearly shows that the sequence identity of the template to the target is not a good descriptor to predict the enrichment obtained by a homology model. Unexpectedly for the InsR kinase, the homology model based upon the structure co-crystallized with an ATP analog (1ir3) yields no enrichment, whereas the model based upon an apo structure (1irk) performs comparably to the 1jqh and 1p4o structures. It is worth mentioning that the natural choice for an IGF-1R template is 1ir3, since 1irk is an unbound structure and all the other templates are much farther away in the sequence space. As for the 1jqh and 1p4o structures, the homology models perform significantly worse than aew in the top 5% of the database, which originates from an induced-fit effect of NVP-AEW541 upon binding (as shown above).

Enrichment curves for the two homology models built upon the insulin receptor kinases. The enrichment curve for the aew structure is shown for comparison

Enrichment curves for the four homology models built upon the fibroblast growth factor receptor kinases. The enrichment curve for the aew structure is shown for comparison
Figure 6 and Table 1 show for the IGF-1R structures and the homology models the values for the area under the ROC curve and the enrichment factors at 2% of the database screened. From the perspective of HTD of large compound libraries, we consider the latter to be more important than the former. In terms of enrichment factor (@2%), the aew outperforms all the other receptors, including the two other IGF-1R structures, which is due to induced-fit effects (see above). The picture differs for the area under the ROC curve, where five homology models perform slightly better than the aew structure. Overall, a large range of performance can be observed, which shows that the nature of the bound ligand can significantly bias the results of docking. The enrichment factors range from slightly better than random (1.5 for the receptor built upon the 1fgi template) to 7.9, which is obtained for a homology model based upon 1y57. Although the latter enrichment is significantly worse than that obtained by aew (12.6), it is better than those obtained by the two other IGF-1R structures (6.2 and 6.3). The values for the area under the ROC curve range from random (0.50) to fair enrichment (0.76). We note that 8 out of the 21 homology models (38%) yield ROC values above 0.70, and that 3 out of the 4 homology models that yield the worst ROC values were co-crystallized with an ATP analog (1pkg, 1ir3, and 1mp8).

ROC values and enrichment factors at 2% of the database screened for the 21 homology models (black) and for the 3 IGF-1R crystal structure (red). The homology models built upon structures co-crystallized with an ATP analog are also indicated
Figure 7 represents the enrichment at 2% of the database screened and the area under the ROC curve as a function of the template-to-target binding-site sequence identity. No correlation can be observed, which is consistent with previous studies [10, 11]. Using the kinase-domain sequence identity leads to the same conclusion (data not shown). This result can be explained by different factors. First, the receptor is kept rigid during docking, whereas the true structure of the crystal structure of the target depends on the bound ligand. Therefore, since the crystal structure is not entirely correct for some of the binders, a higher sequence similarity with the target protein will not necessarily translate into an improved enrichment. Very recently, the influence of protein structure on the successful reproduction of known ligand poses by Glide was investigated using 20 different CDK2 structures and a set of 340 binders [31]. The rate of correct pose reproduction was found to range from 16% to 82%. This result should also translate into a large range of enrichment results in virtual screening. In a docking study based on CDK2 and neuraminidase, it has been shown that enrichment is significantly higher for the actives whose binding modes are predicted correctly [16]. If various crystal structures can yield very different enrichments, the same result can be expected for homology models. Second, like most of the scoring functions, the Glide scoring function depends relatively strongly on the exact protein-ligand conformation, i.e., small changes in the conformation can result in large changes in the binding scores. Therefore, docking into two homology models with a high sequence similarity to the target and small conformational differences can yield significantly different compound rankings.

Enrichment factor at 2% of the database screened and area under the ROC curve as a function of the template-to-target binding-site sequence identity. The results for the homology models and IGF-1R structures are shown in black and red, respectively
A structural analysis was carried out and is shown in Figs. 8 and 9. Performance does not correlate with the binding-site RMSD from the aew structure, although the homology models that perform worst have an RMSD above 2 Å (Fig. 8). Figure 9 represents the enrichment at 2% of the database screened and the area under the ROC curve as a function of the binding-site volume. The volume was computed using the program voidoo [32]. The three homology models that yield the worst performance have the smallest binding-site volume. This fact can be used as a criterion to discard homology models, e.g. those with a binding-site volume below 600 Å3. This result is consistent with a very recent study based on CDK2, where it was found that the main determinants of the ability of Glide to successfully reproduce known ligand binding modes are the volume of the binding site and the exact orientation of the residues forming the binding site [31].

Enrichment factor at 2% of the database screened and area under the ROC curve as a function of the heavy-atom RMSD from the aew structure. The results for the homology models and IGF-1R structures are shown in black and red, respectively

Enrichment factor at 2% of the database screened and area under the ROC curve as a function of the ATP binding site volume. The results for the homology models and IGF-1R structures are shown in black and red, respectively
HTD with homology models can yield a substantial enrichment of known actives against a background of decoys. Indeed, some homology models yield an area under the ROC curve that is comparable to those obtained by the crystal structures. Nevertheless, the results also show that a wide range of performance can be expected with homology models, which poses the question regarding the selection of the template. Although the volume of the binding site can be used to remove a template that would yield a very poor enrichment, there is a priori no way to find out which model will perform best. As a result, a safe choice might be to do HTD with an ensemble of models. Docking to multiple receptors is less likely to produce very poor results than docking to a single cavity. It has been shown that docking against multiple receptor conformations yields improved enrichment versus docking against a single conformation [33, 34]. Similarly to the consensus scoring function [35], a consensus cavity scoring approach has been proposed to improve hit rates [36].
Here, we make use of the homology models to assess the performance of ensemble docking. When HTD is performed against multiple receptor conformations, a post-processing step is carried out, where the results of the HTD experiment against each conformation are merged by keeping for each compound the best rank according to the Glide score. Therefore, the size of this ranked list corresponds to that of a HTD against a single receptor. We will refer to here as the merge-and-shrink procedure, which has been used recently [34]. We also used the scores to combine the results of multiple receptors and we obtained very similar results (data not shown). In order to identify the multiple receptor conformations, the 21 binding sites were clustered using XCluster [37] accessible through the Maestro interface and the conformation closest to the centroid was kept as a representative for each cluster. The enrichment at 2% of the database screened and the area under the ROC curve are represented in Fig. 10 as a function of the number of clusters. When all the homology models are grouped into two clusters, the enrichment factor (@2%) obtained by the merging-and-shrinking is 5.5, which is very similar to those obtained by the two representatives (5.6 and 5.5). On the other hand, this result is much better than that one would have obtained by choosing 1ir3 as a template, which is the natural choice for an IGF-1R template. Figure 10 also shows that using more than two receptor conformations does not improve the enrichment value. Indeed, the enrichment factor obtained by the merge-and-shrink procedure is 4.3 when the homology models are grouped into three clusters. The decrease in performance is due to the fact the enrichment obtained by the 1ywn template, which is the representative of the third cluster when going from two to three clusters, is only 2.6. The inactive ligands that receive low docking scores in the 1ywn receptor are “carried over” with the merge-and-shrink procedure, thus increasing the rate of false positives and worsening the performance. On the contrary, using more and more conformations improves the ROC value (Fig. 10). The ROC value obtained by the merging-and-shrinking applied to four structures is 0.74, which is very close to the value obtained by the best homology model (0.76). Furthermore, the merging-and-shrinking always yields a ROC value that is better than those obtained by the single receptors. This result can be explained by the fact that the merge-and-shrink procedure improves the docking scores of the active compounds. When applied to three conformations, the average Glide score for the binders is −7.1, whereas the corresponding value for the receptors is −6.5, −5.9, and −6.8, respectively. It is instructive to compare the binders that are ranked in the top 2% scored poses by the homology models. As an example, we consider the representatives of the three clusters obtained by grouping all the homology models. These three structures have only three actives in common in the top 2% and, therefore, it is likely that ensemble docking should improve the diversity of the hits.

Enrichment factor at 2% of the database screened and area under the ROC curve as a function of the number of clusters. The results obtained by the merging-shrinking procedure and by the individual homology models are shown in black and colors, respectively
Conclusions
In this study, 21 homology models of the IGF-1R kinase were built to assess their ability to discriminate active from inactive compounds in HTD. More than 600 known IGF-1R actives were pooled with 300,000 compounds, which were considered to be inactive. All compounds were docked into the ATP binding site of the 21 homology models and the three IGF-1R crystal structures, and the performance ranks of the known actives with respect to the remaining pool were converted into enrichment and ROC plots. We have found that the homology models yield a large range of performance from random to as good as those obtained by two of the three crystal structures of the target protein. We have even observed that different homology models based on templates with the same sequence identity yield very different enrichments. In terms of enrichment factors at 2% of the database screened, the results range from an 1.5-fold to an 7.6-fold enrichment compared with random screening. As a basis of comparison, the enrichment factors obtained by the IGF-1R structures range from an 6.2-fold to a 12.6-fold enrichment. Furthermore, the best crystal structure outperforms the best homology model and we have also shown that this result originates from an induced-fit effect. In terms of ROC values, the performance for the homology models and the IGF-1R crystal structures ranges between 0.50 and 0.76, and between 0.67 and 0.73, respectively.
Based on a structural analysis of the results, we can formulate a few recommendations for HTD on kinases. First, we have seen that the receptors with the smallest binding site perform worst and, therefore, the volume of the ATP binding site can be used as a criterion to discard a template. On the other hand, Fig. 9 also shows that the performance of the homology models does not correlate with the size of the binding site for values above 600 Å3. It is therefore likely that some regions of the binding site are more important than others. Second, the results have shown that the homology models based upon a structure co-crystallized with an ATP analog perform poorly and should not be used. Third, we have proposed to combine the compound rankings of multiple receptor conformations as a way to take into account receptor flexibility. Further work is needed to determine the optimal way of choosing these conformations. At present, a reasonable choice is to select conformations with various shapes and sizes. It should be kept in mind that allowing conformational flexibility might increase the rate of false positives, since inactive compounds could be docked with a better score. At the very least, this approach should avoid the situation where HTD yields no enrichment.
Overall, this study shows that the rigidity of the receptor is an important limitation in HTD and that the selection of the template(s) is a key aspect in comparative modeling. Furthermore, our results are consistent with previous studies based on other docking algorithms and targets. Therefore, it is unlikely that the protein target and docking method used in this work have a significant bias on the conclusions. From the perspective of HTD, it may be difficult to choose which structure to use. If a compound is known to bind the target protein, a strategy would be to use this information to validate the homology models. In this respect, an algorithm that makes use of the known binding mode of a binder during the homology modeling process has been developed [38].
References
Jacobson MP, Sali A (2004) Ann Reports Med Chem 39:259–276
Hillisch A, Pineda LP, Hilgenfeld R (2004) Drug Discovery Today 9:659–669
Bissantz C, Bernard P, Hilbert M, Rognan D (2003) Proteins 50:5–25
Varady J, Wu X, Fang X, Min J, Hu Z, Levant B, Wang S (2003) J Med Chem 46:4377–4392
Evers A, Klabunde T (2005) J Med Chem 48:1088–1097
Salo OMH, Raitio KH, Savinainen JR, Nevalainen T, Lahtela-Kakkonen M, Laitinen JT, Järvinen T, Poso A (2005) J Med Chem 48:7166–7171
Vangrevelinghe E, Zimmermann K, Schoepfer J, Portmann R, Fabbro D, Furet P (2003) J Med Chem 46:2656–2662
McGovern SL, Shoichet BK (2003) J Med Chem 46:2895–2907
Diller DJ, Li R (2003) J Med Chem 46:4638–4647
Oshiro C, Bradley EK, Eksterowicz J, Evensen E, Lamb ML, Lanctot K, Putta S, Stanton R, Grootenhuis PDJ (2004) J Med Chem 47:764–767
Kairys V, Fernandes MX, Gilson MK (2006) J Chem Inf Model 46:365–379
Kenyon V, Chorny I, Carvajal WI, Holman TR, Jacobson MP (2006) J Med Chem 49:1356–1363
Blume-Jensen P, Hunter T (2001) Nature 411:355–365
Wagman AS, Nuss JM (2001) Curr Pharm Des 7:417–450
Adcock IM, Chung KF, Caramori G, Ito K (2006) Eur J Pharmacol 533:118–132
Verdonk ML, Berdini V, Hartshorn MJ, Mooij WTM, Murray CW, Taylor RD, Watson P (2004) J Chem Inf Comput Sci 44:793–806
Muegge I, Enyedy IJ (2004) Curr Med Chem 11:693–707
Chuaqui C, Deng Z, Singh J (2005) J Med Chem 48:121–133
Sims PA, Wong CF, McCammon JA (2003) J Med Chem:46:3314–3325
Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S (2002) Science 298:1912–1934
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) Nucleic Acids Research 28:235–242
Vieth M, Higgs RE, Robertson DH, Shapiro M, Gragg EA, Hemmerle H (2004) Biochim Biophys Acta 1697:243–257
Glide, version 4.0, Schrödinger, LLC, New York, NY, 2005
Garcia-Echeverria C, Pearson MA, Marti A, Meyer T, Mestan J, Zimmermann J, Gao JP, Brueggen J, Capraro HG, Cozens R, Evans DB, Fabbro D, Furet P, Porta DG, Liebetanz J, Martiny-Baron G, Ruetz S, Hofmann F (2004) Cancer Cell 5:231–239
Prime, version 1.5, Schrödinger, LLC, New York, NY, 2005
Maestro, version 7.5, Schrödinger, LLC, New York, NY, 2006
Sadowski J, Rudolph C, Gasteiger J (1990) Tetrahedron Comp Method 3:537–547
LigPrep, version 2.0, Schrödinger, LLC, New York, NY, 2005
Pipeline Pilot 5.0; Scitegic, Inc.
Witten IH, Frank E (1999) Data mining: practical machine learning tools and techniques with java implementations. Morgan Kaufmann Publishers, New York
Thomas MP, McInnes C, Fischer PM (2006) J Med Chem 49:92–104
Kleywegt GJ, Jones TA (1994) Acta Cryst D50:178–185
Ferrari AM, Wei BQ, Constantino L, Shoichet BK (2004) J Med Chem 47:5076–5084
Cavasotto CN, Abagyan RA (2004) J Mol Biol 337:209–225
Charifson PS, Corkery JJ, Murcko MA, Walters WP (1999) J Med Chem 42:5100–5109
Barril X, Morley SD (2005) J Med Chem 48:4432–4443
MacroModel XCluster, version 9.1, Schrödinger, LLC, New York, NY, 2005
Evers A, Gohlke H, Klebe G (2003) J Mol Biol 334:327–345
Acknowledgements
We thank Dr Nathan Brown for careful reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ferrara, P., Jacoby, E. Evaluation of the utility of homology models in high throughput docking. J Mol Model 13, 897–905 (2007). https://doi.org/10.1007/s00894-007-0207-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-007-0207-6