
Abstract
An extracellular, halophilic, alkalithermophilic serine protease from the halo-alkaliphilic Alkalibacillus sp. NM-Da2 was purified to homogeneity by ethanol precipitation and anion-exchange chromatography. The purified protease was a monomeric enzyme with an approximate molecular mass of 35 kDa and exhibited maximal activity at 2.7 M NaCl, pH55 °C 9 and 56 °C. The protease showed great temperature stability, retaining greater than 80 % of initial activity after 2 h incubation at 55 °C. The protease was also extremely pH tolerant, retaining 80 % of initial activity at pH55 °C 10.5 after 30 min incubation. Protease hydrolyzed complex substrates, displaying activity on yeast extract, tryptone, casein, gelatin and peptone. Protease activity was inhibited at casein concentrations greater than 1.2 mg/mL. The enzyme was stable and active in 40 % (v/v) solutions of isopropanol, ethanol and benzene and was stable in the presence of the polysorbate surfactant Tween 80. Activity was stimulated with the oxidizing agent hydrogen peroxide. Inhibition with phenyl methylsulfonylfluoride indicates it is a serine protease. Synthetic saline wastewater treated with the protease showed 50 % protein removal after 5 h. Being halophilic, alkaliphilic and thermophilic, in addition to being resistant to organic solvents, this protease has potential for various applications in biotechnological and pharmaceutical industries.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Proteases are a group of hydrolytic enzymes that cleave peptide bonds and degrade proteins into smaller peptides and amino acids. Subsequently they play a prominent role in various physiological processes in all domains of life. The application of proteases in detergent, silk, leather, bakery, soy processing and meat tenderizing industries is well documented (Delgado-Garcia et al. 2012; Gupta et al. 2002; Karbalaei-Heidari et al. 2013). Proteases also have numerous medical and pharmaceutical applications, such as synthesis of antibodies, hormones and biomedical peptides (Chanalia et al. 2011). Operation of proteases in ionic liquids and organic solvents is a developing area in biochemistry and biotechnology since they can catalyze synthetic and transesterification reactions with reasonable stereospecificity at low water activity (Ogino and Ishikawa 2001). All of the above mentioned applications demand that proteases be stable to salts, organic solvents, alkaline pH and high temperature.
A problem commonly encountered with using enzymes for industrial biocatalysis is their inherent stability to the conditions used. Extremophiles can play a major role in addressing this issue. Extremophiles are organisms that survive, grow and thrive under environmental conditions considered to be extreme by human standards, such as high salinity, alkaline or acid pH, high or low temperature and high concentrations of organic solvents (Mesbah and Wiegel 2012). These adaptations allowed enzymes from extremophiles, called extremozymes, to expand the ranges for optimal enzyme performance thereby enabling biocatalysis under various conditions commonly encountered in industrial processes (Sarmiento et al. 2015).
Halophiles are the most promising source of enzymes stable in salts and organic solvents. Halophiles inhabit hypersaline environments where salinity often reaches saturation, and accumulate large concentrations of salts and/or organic compatible solutes in their cells as an adaptation (DasSarma and DasSarma 2015; Oren 2010). High salt concentration lowers water activity, therefore both the extra- and intracellular conditions of halophiles resembles nonaqueous systems. Numerous halophilic proteases have been purified and described (Yin et al. 2015). These proteases remain active at sodium chloride concentrations greater than 1.5 M (and up to 2.5 M), and most of them are tolerant to a pH of 5–8 and temperature of 40–45 °C.
Known alkaline proteases generally show maximal activity at pH 9–10 and 50–60 °C (Borkar 2015). Their activity, however, is often greatly reduced at salt concentrations greater than 0.5 M, thereby reducing their use in applications requiring high salt concentration and low water activity. Thermophilic proteases are stable and active at temperatures between 60 and 80 °C and are generally resistant to alkaline and acid pH, but as far as described, they are not stable at salt concentrations greater than 1 M (Raddadi et al. 2015; Synowiecki 2015).
Despite the numerous advantages of extremozymes, and the great advances in extremozyme research over the past two decades (Borkar 2015; DasSarma and DasSarma 2015; Sarmiento et al. 2015), available extremophilic biocatalysts remain limited. In addition, reports on extremozymes capable of catalyzing reactions in the presence of more than two extreme conditions, such as high salt, alkaline pH and high temperature, are scarce (Sarmiento et al. 2015). Poly-extremozymes have greater versatility to the combination of harsh conditions usually encountered in industrial applications, such as low water activity, high concentration of organic solvents, alkaline pH and high temperature. Therefore, the isolation and purification of poly-extremozymes is essential to expand the existing industrial enzyme toolbox. The present work describes the isolation and biochemical characterization of an extremely halophilic, alkaliphilic and moderate thermophilic protease produced by Alkalibacillus sp. NM-Da2 that was isolated from the soda lakes of the Wadi An Natrun, Egypt. This protease is stable in the presence of organic solvents and surfactants, and has potential for application in different industrial processes.
Materials and methods
Organism and culture conditions
Protease secreting strains were isolated from mixed water–sediment samples collected from the largest lakes of the Wadi An Natrun in North–Western Egypt as described (Mesbah and Wiegel 2014). Approximately 5 g (wet weight) was inoculated into enrichment medium consisting of, in g/L NaCl, 100, Na2CO3, 20, KH2PO4, 1, MnCl2, 0.008, FeCl2, 0.05, KCl, 0.5, MgCl2, 0.5, yeast extract, 5, peptone, 5. Enrichment cultures were incubated at 50 °C and pH55 °C 9 for 72 h [superscript after the pH indicates temperature at which the pH was measured and the pH electrode calibrated. For details, see Wiegel (1998)]. Pure isolates were obtained by repetitive dilution to extinction in the same culture medium supplemented with 1 % (w/v) agar. Isolates were screened for extracellular protease activity by protease assay using the Folin–Ciocalteu reagent. Isolate NM–Da2 displayed proteolytic activity, and was selected for detailed study.
Taxonomic position of isolate NM-Da2 was identified by 16S rRNA sequence comparison. Genomic DNA was extracted by the CTAB/NaCl method of Wilson (Wilson 1997). The 16S rRNA gene was amplified by polymerase chain reaction with universal primers 27F (5′-AGA GTT TGA TCM TGG CTC AG-3′) and 1492R (5′-GGT TAC CTT GTT ACG ACT T-3′) and DreamTaq™ green PCR master mix (ThermoFisher Scientific). Amplified products were sequenced using a 3730 × l capillary DNA analyzer (Applied Biosystems) operated at the Georgia Genomics Facility (University of Georgia, Athens, GA USA). Phylogenetic analysis was performed as described (Mesbah and Wiegel 2014).
The same culture medium described above was used for enzyme production. One hundred milliliters of culture were grown in 250 mL Erlenmeyer flasks at 50 °C with shaking (115 rpm) in an Excella™ E24R temperature controlled benchtop shaker (Eppendorf™). After 5 days of incubation, the culture was centrifuged at 3000×g for 30 min at 4 °C and the supernatant was used for protease purification.
Protease purification
Proteins in the cell free supernatant were precipitated by addition of 0.8 volumes of ice cold absolute ethanol and incubation at -20 °C overnight. Precipitated proteins were collected by filtration using filter paper (Whatman #1). The precipitate was dissolved in a minimal amount of 50 mM glycine NaOH, pH 9 (~50 mL). Suspended protein was dialyzed against 2 L of the same buffer overnight, with one buffer change to reduce residual ethanol and salts.
Dialyzed protein was filtered through a 0.2 μm pore. Proteins were separated by anion-exchange chromatography using an Econo™ gradient pump equipped with a gradient mixer and EM-1 Econo™ UV monitor (BioRad, USA) with a Q-Sepharose FF column (10 × 1.6 cm, GE Healthcare). The column was equilibrated with 50 mM Tris.Cl, pH 9.0. Bound proteins were eluted with a linear gradient of 0–2 M NaCl in Tris.Cl, pH 9.0 at a flow rate of 2.5 mL/min. Protein content (absorbance at 254 nm) and protease activity was assessed for eluted fractions. Fractions showing highest protease activity were pooled, desalted by dialysis against 50 mM glycine NaOH, pH 9, and concentrated by ultrafiltration through a cellulose membrane with 10 kDa cutoff (Amicon®). The concentrated protein was used for further biochemical characterizations.
Protease assay
Protease activity was determined by measuring tyrosine liberated during casein hydrolysis using the Folin–Ciocalteu reagent as described by Anson (1938). The assay mixture (200 μL final volume) consisted of purified enzyme (2–3 μg), 2.6 M NaCl and 50 mM Tris.Cl, pH55 °C 9. The reaction was started with the addition of casein (LabM, UK) to a final concentration of 0.1 %, w/v. Reaction mixtures were incubated at 55 °C for 30 min. The reaction was stopped by the addition of 250 μL of 110 mM trichloroacetic acid (TCA) solution and centrifugation at 8000×g for 5 min. To 400 μL of supernatant, 800 μL of 500 mM Na2CO3 solution and 200 µL of 0.5 N Folin–Ciocalteu reagent were added. The mixture was mixed thoroughly and incubated at room temperature for 15 min. Color developed was then immediately measured at 660 nm. All assays were performed in triplicate. Standard deviation and standard error were calculated for all assays. A negative control (without protein) was run for all assays to correct for nonspecific color development and nonspecific hydrolysis of substrate. One unit of protease activity was defined as the amount of enzyme yielding 1 μmol of tyrosine per minute at pH55 °C 9.0, 55 °C in the presence of 2.6 M NaCl. Specific activity of protein is expressed as the units of enzyme activity per milligram protein. Protein concentration was measured by the method of Bradford (1976), using bovine serum albumin (BSA) as standard.
Polyacrylamide gel electrophoresis and zymography
Molecular weight and purity of the enzyme was estimated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) in a discontinuous system as described by Laemmli (1970). Samples were boiled for 3 min in Laemmli sample buffer then loaded onto 10 % polyacrylamide gels. After electrophoresis, the gels were stained with the SERVA silver staining kit (Serva Gmbh, Heidelberg, Germany). Molecular weight of the enzyme was estimated using an EZ-Run™ Rec protein ladder (Fisher BioReagents).
Zymography was performed on SDS-PAGE by the method described by Schmidt et al. (1988) with modifications. Polyacrylamide gels (10 %) were copolymerized with 0.1 % (w/v) gelatin. Samples were mixed with Laemmli sample buffer without heat denaturation and electrophoresed at 125 V for 45 min. After electrophoresis, gels were washed once in 50 mM Tris.Cl, pH 9, then immersed in 50 mM Tris.Cl, pHRT 9 containing 2.5 % (v/v) Triton X-100 and incubated at room temperature with shaking for 30 min to remove SDS. Triton X-100 was removed by washing gels three times in 50 mM Tris.Cl, pHRT 9 (10 min each) at room temperature. Gels were then incubated in 50 mM Tris.Cl, pH55 °C 9 containing 2.6 M NaCl at 55 °C with gentle shaking (50 rpm) for 30 min. Gels were washed with distilled water for 1 min, then stained with a 0.05 % (w/v) solution of Coomassie blue R (Fisher BioReagents) in 10 % acetic acid, 50 % methanol, for 30 min. Finally, gels were destained (7 % acetic acid, 5 % methanol) for 2 h. Proteolytic activity was demonstrated as a clear band of lysis against a blue background.
Biochemical characterization of purified protease
Effect of salt, pH and temperature
Effect of [Na+] on protease activity was determined by measuring the rate of casein degradation by the purified enzyme at NaCl concentrations from 0 to 3.4 M, at pH55 °C 9 and 55 °C.
To determine effect of pH on protease, protease activity was tested over a pH55 °C range 7–12 in buffer consisting of 50 mM Tris.Cl, 50 mM glycine, 50 mM phosphate and 2.6 M NaCl at 55 °C.
Protease activity was determined at temperatures 30–75 °C at pH55 °C 9 in the presence of 2.6 M NaCl. Heat stability of the purified protease was determined after incubation of the enzyme at 45, 50, 55, 60 and 65 °C for 24 h. Samples were drawn and residual protease activity was determined by standard assay at 55 °C, pH55 °C 9, and 2.6 M NaCl. Enzyme that had not been incubated served as the 100 % control.
Substrate specificity and effect of substrate concentrations on protease activity
Substrate specificity was determined under standard assay conditions using different protein substrates including gelatin (Loba Chemie, India), peptone (Oxoid), tryptone (LabM, UK), yeast extract (LabM, UK), skimmed milk (LabM, UK), human hemoglobin (Sigma-Aldrich), BSA (Fluka), casein (LabM, UK) and wheat gluten (Sigma-Aldrich). Substrates were added to a final concentration of 0.1 % (w/v).
Effect of substrate concentration on reaction velocity was assayed at pH55 °C 9, 2.6 M NaCl and 55 °C with casein as substrate. The concentration of casein was increased from 0 to 3 mg/mL.
Effect of organic solvents
Stability and activity of the enzyme in the presence of organic solvents was evaluated by pre-incubating the enzyme at 55 °C, 2.6 M NaCl and pH55 °C 9 with 20 or 40 % (v/v) each of ethanol, methanol, butanol, isopropanol, benzene, chloroform and hexane for 30 min. Protease activity was then measured by the standard assay after addition of substrate. Activity in the absence of organic solvents was taken as 100 %.
Effect of surfactants and detergents
Proteolytic activity was measured by the standard assay in the presence of 1 and 5 % (v/v) final concentration of different surfactants, such as sodium dodecyl sulfate (SDS) (Fluka), Tween 20 (Sigma-Aldrich), Tween 80 (Oxford Chemicals), Triton X-100 (Fluka) and the oxidizing agent H2O2.
The purified enzyme was pre-incubated with each surfactant or chemical agent at 50 °C, pH55 °C 9.0 in the presence of 2.6 M NaCl in the absence of substrate for 30 min. The reaction was started with the addition of substrate. After 30 min of incubation, the reaction was terminated by the addition of TCA and liberated tyrosine was measured as described above. No-enzyme controls were run for all assays to correct for nonspecific color development and nonspecific hydrolysis of substrate.
Effect of enzyme inhibitors
Proteolytic activity was measured by the standard assay in the presence of different protease inhibitors, such as phenyl methylsulfonylfluoride (PMSF), dithiothreitol (DTT), ethylene diamine tetraacetic acid (EDTA), urea and β-mercaptoethanol with final concentrations 2 and 5 mM. All inhibitors were purchased from Sigma-Aldrich.
The purified enzyme was pre-incubated with each inhibitor at 50 °C, pH55 °C 9.0 in the presence of 2.6 M NaCl and the absence of substrate for 30 min. The reaction was started with the addition of substrate. After 30 min of incubation, the reaction was terminated by the addition of TCA and liberated tyrosine was measured as described above. No-enzyme controls were run for all assays. Assays were performed in triplicate.
Effect of metal salts
The effect of metal salts on the purified protease was tested by the standard protease assay in the presence of different ions to identify potential activators or inhibitors.
The effect of 2 and 5 mM (final concentration) of the chloride salts of Na+, Ba2+, Mn2+, Cu2+, Hg2+, Fe2+, Zn2+, Ca2+, Mg2+, K+, Sr2+, Cs2+, NH4+ and Ag+ on protease activity was determined at 55 °C and pH55 °C 9.0.
Bioremediation application
Synthetic wastewater used was composed of, in mg/L: beef extract, 50; peptone 80; urea, 30; K2HPO4, 28; CaCl2, 4; MgSO4, 2 and 2.6 M NaCl. pH55 °C was adjusted to 9. Purified protease was added with different amounts (1, 2, and 3 % w/v) and degradation was performed at 55 °C. The reactions were shaken gently (25 rpm) to ensure uniform mixing and samples were collected at time intervals for analysis of total protein content. Total protein content was measured by the Bradford assay (Bradford 1976).
Results and discussion
Isolation of strain NM-Da2 and production of protease
Strain NM-Da2 was isolated from Lake Abo-Dawood, Wadi An Natrun, Egypt in enrichment medium containing 0.5 % w/v each peptone and yeast extract at pH55 °C 9 in the presence of 2.6 M NaCl. Strain NM-Da2 was Gram-staining positive, aerobic and rod-shaped. Colonies were yellow-white in color. Phylogenetic analysis based on 16S rRNA comparison showed that the isolate belongs to the genus Alkalibacillus (Fig. 1). The 16S rRNA sequence from stain NM-Da2 was deposited in GenBank under accession number KU740034.

Neighbor joining tree based on 16S rRNA sequences showing phylogenetic position of strain NM-Da2 in relation to type species of the genus Alkalibacillus. The tree was rooted with the 16S rRNA sequence of Bacillus subtilis DSM 10T as outgroup. GenBank accession numbers are shown in parentheses. Bootstrap values based on 100 replicates are indicated at nodes. Bar 1 nucleotide substitution per 100 bases
Strain NM-Da2 produced an extracellular protease in liquid medium containing 0.5 % w/v each yeast extract and peptone. The observed protease activity in the culture was dependent on the growth phase, measuring 120 U during early exponential phase and reaching a maximum (1380 U) in the stationary growth phase. In the absence of yeast extract and peptone both growth and protease activity were negligible.
Protease purification
Extracellular protease from Alkalibacillus sp. NM- Da2 was precipitated from 600 mL of culture supernatant by ethanol precipitation and anion-exchange chromatography leading to 11.3 fold purification and a specific activity of 74 U/mg (Table 1).
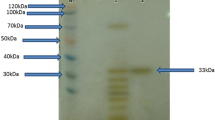
The purified protease migrated as a single band on SDS-PAGE with a molecular weight of approximately 35 kDa (Fig. 2a). The purified enzyme also migrated as a single band on a non-denaturing gel, indicating it is monomeric. Gelatin zymography confirmed the proteolytic nature of the enzyme (Fig. 2b). The culture supernatant also showed a single zone of hydrolysis on zymography, indicating that Alkalibacillus sp. NM-Da2 does not produce extracellular proteases with different molecular weights.

SDS-PAGE of purified protease. a Assessment of the homogeneity and molecular weight of the protease by 10 % SDS-PAGE. Lanes 1–3 represent: molecular weight marker, ethanol precipitated protein, purified protein. b Zymogram of purified protease. Proteolytic activity is manifested by a clear band on a dark background
The protease from Alkalibacillus sp. NM-Da2 was of similar molecular weight to halophilic proteases isolated from Bacillus sp. EMB9 (29 kDa) (Sinha and Khare 2013), Natronolimnobius innermongolicus WN18 (47 kDa) (Selim et al. 2014), Alkalibacillus sp. NM-Fa4 (20 kDa) (Mesbah and Wiegel 2014), Bacillus subtilis AP-MSU (18.3 kDa) (Maruthiah et al. 2013) and Bacillus cereus SIU1 (22 kDa) (Singh et al. 2012). However, it was smaller than halophilic proteases isolated from Bacillus sp. SM2014 (71 kDa) (Jain et al. 2012) and Bacillus sp. CY7 (97 kDa) (Arabaci et al. 2013).
Characterization of the purified protease from Alkalibacillus sp. NM-Da2
Effect of salt, pH and temperature on protease activity
The protease has a narrow [Na+] range for activity, showing maximal activity at 2.7 M NaCl (at pH55 °C 9 and 55 °C) (Fig. 3). The purified enzyme retained 38 % of its activity at 3.1 M NaCl and showed only 16 % of maximal activity in the absence of NaCl (Fig. 3). The protease was alkaliphilic, with maximal activity at pH55 °C 9, and retained 80 % of activity at pH55 °C 10.5 (Fig. 4). Protease retained only 6 % of maximal activity at pH55 °C 7, and lost more than 70 % of its activity at pH55 °C 11.5.

Effect of NaCl on protease activity. Assays were performed at pH55 °C 9 and 55 °C. Activity at 2.7 M NaCl was taken as 100 %, and was equivalent to 93 U/mg. Values represent the mean of three independent assays, bars standard error

Effect of pH55 °C on protease activity at 55 °C and 2.6 M NaCl. Activity at pH55 °C 9 was taken as 100 % and was equivalent to 90 U/mg. Each value represents mean of three independent assays, bars denote standard error
The temperature range for activity was between 30 and 75 °C (at pH55 °C 9 in the presence of 2.6 M NaCl), with a narrow maximal peak at 56 °C. The enzyme retained approximately 50 % of maximal activity at 75 °C (Fig. 5a).

Effect of temperature on protease (A) activity and (B) stability at pH55 °C 9 and 2.6 M NaCl. (A) Effect of temperature on protease activity. Activity at 56 °C was taken as 100 %, and was equivalent to 94 U/mg. (B) Temperature stability of protease. Protein was incubated at 45 °C (filled circle), 50 °C (open circle), 55 °C (filled diamond), 60 °C (filled square) and 65 °C (open square) at pH 9. Activity was assayed at defined time intervals. Each value represents mean of three independent assays, bars standard error
The protease was heat stable, retaining over 80 % of its initial activity after 180 min incubation at 45, 50 and 55 °C (Fig. 5b), and retained over 50 % of activity after 24 h. The enzyme retained 73 and 67 % of initial activity after incubation at 60 and 65 °C, respectively, for 1 h, and lost activity after 3 h of incubation.
Stability at high ionic strength in addition to elevated temperature and extreme alkaline pH makes the protease from Alkalibacillus sp. NM-Da2 a good candidate for use in different industrial applications (Yin et al. 2015). Alkaline, salt- and temperature stable proteases have commercial uses as components in laundry detergents, photographic gelatin hydrolysis and leather dehairing (Sarethy et al. 2011). Salt- and thermostable alkaline proteases have recently found new applications such as the cleaning of ultrafiltration membranes used by chemical and pharmaceutical industries. In addition, halophilic, alkali- and thermostable proteases have great potential for scientific research and discovery, their selective peptide bond cleavage can be used in the elucidation of structure–function relationship, peptide synthesis, sequencing of proteins and production of amino acids (Jisha et al. 2013).
Few studies report on proteases active under more than two extreme conditions. Cysteine protease AbCP, produced by Alkalibacillus sp. NM-Fa4 isolated from the Wadi An Natrun, had maximal activity at pH45 °C 9.5, 50–52 °C and 1 M NaCl, but lost 40 % of activity at NaCl concentrations greater than 2 M (Mesbah and Wiegel 2014). The thermostable, alkaliphilic, halophilic protease from the archaeon Natronolimnobius innermongolicus WN18, also isolated from the Wadi An Natrun, showed maximal activity at 60 °C and pH 9–10 in the presence of 2.5 M NaCl (Selim et al. 2014). The Bacillus sp. CY7 extracellular protease has maximal activity at 70 °C and pH 11, but was only active at 0–0.5 M NaCl (Arabaci et al. 2013). The Bacillus sp. EMB 9 serine protease has maximal activity at pH 9 and 55◦C, but could only tolerate 0.2 M NaCl (Sinha and Khare 2013). A serine protease produced by Bacillus subtilis AP-MSU 6 exhibited maximal activity at pH 9, 0.5 M NaCl and 40 °C, but rapidly lost activity at NaCl concentrations larger than 0.5 M and temperatures greater than 45 °C (Maruthiah et al. 2013). The extracellular protease produced by Bacillus cereus S1U1 is active at pH 9, 45–55 °C but retained only 67 % of its initial activity in the presence of 1.2 M NaCl (Singh et al. 2012). Bacillus sp. SM2014 produces an alkaliphilic, thermophilic protease with maximal activity at pH 10–11, 60–70 °C, 0.2 M NaCl, and retained 93 % of activity at 1.5 M NaCl (Jain et al. 2012).
Substrate specificity of protease from Alkalibacillus sp. NM-Da2
The protease had high hydrolytic activities against complex substrates yeast extract, casein, tryptone, peptone and gelatin (Table 2), but weak activity against skimmed milk, hemoglobin, BSA and wheat gluten.
With casein as substrate, the protease displayed non-Michealis–Menten kinetics, with substrate inhibition above 1.2 mg/mL casein and complete inhibition at 2.5 mg/L (Fig. 6).

Effect of casein concentration on the reaction velocity of protease. Assays were performed at 55 °C, pH55 °C 9 and 2.6 M NaCl. Each value represents the mean of three assays, bars denote standard error
The substrate utilization spectrum was narrow compared with other halophilic, alkalithermophilic proteases (Arabaci et al. 2013; Jain et al. 2012; Joshi and Satyanarayana 2013; Maruthiah et al. 2013; Selim et al. 2014; Singh et al. 2012; Sinha and Khare 2013). This may limit the application of the protease in industrial applications, but it can find application in processes that involve dissolved organic matter, such as bioremediation.
Effect of organic solvents on protease activity
Solvent stability is a characteristic of halophilic enzymes (Delgado-Garcia et al. 2012; Gupta and Khare 2009). Hypersaline solutions are similar to nonaqueous environments as salts reduce water activity of the surrounding medium and compete with other molecules for available water (Oren 2010). The protease was stable and retained more than 70 % of activity in the presence of ethanol, isopropanol and benzene (Table 3). Activity of the protease was enhanced in the presence of 40 % (v/v) isopropanol and lost more than 50 % of its initial activity in the presence of methanol, chloroform, butanol and hexane (Table 3).
Organic, solvent tolerant halophilic enzymes are attractive for environmental applications such as bioremediation of polluted salt marshes and industrial wastewaters contaminated with organic solvents. They are also of significance for biotechnological purposes because they can catalyze peptide and ester synthesis under nonaqueous conditions. There are many advantages to employing proteases in organic as opposed to aqueous media, including higher substrate solubility and the capability to modify substrate specificity of the enzyme by altering the reaction medium (Lee and Dordick 2002).
Solvent stable proteases have been reported. The protease from Bacillus sp. EMB9 is stable in ethanol, methanol, dimethyl sulfoxide, and t-butanol (Sinha and Khare 2013), but lost more than 90 % of activity in the presence of benzene (50 % v/v) and 20 % activity with isopropanol. Bacillus sp. APCMST-RS7 produced a halophilic, organic solvent-tolerant protease that retained full activity in the presence of hexane, methanol, and ethanol, but it was assayed in the presence of 10 and 20 % v/v of solvent. (Maruthiah et al. 2015).
Effect of surfactants and chemical agents
Tween 80 is commonly used as a surfactant in medicated soaps. The presence of an alkalistable protease that is stable in the presence of tween 80 is beneficial, as it can be added to soaps and ointments that are used for the treatment of burns and removal of skin debris (Rosenberg et al. 2004). The protease from Alkalibacillus sp. Da2 was stable in the presence of Tween 80, retaining 81 % of initial activity after 30 min incubation with 1 % v/v of the surfactant (Table 4). The enzyme was sensitive to the surfactants SDS and Tween 20 and the activity was completely inhibited with 5 % (v/v) Triton X-100 (Table 4). Protease activity was enhanced in the presence of the oxidizing agent hydrogen peroxide, showing 124 % of its initial activity after pre-incubation with 5 % (v/v) of the agent (Table 4).
Stimulation of enzyme activity with hydrogen peroxide but inhibition in the presence of other surfactants is noteworthy. It is possible that hydrogen peroxide interacts with the enzyme either at the active site or any other sites in a manner that causes enhanced activity and/or stability. Stimulation of protease activity in the presence of hydrogen peroxide has been reported for the protease AbCP (Mesbah and Wiegel 2014) and Bacillus clausii I-52 (Joo et al. 2003).
The protease was sensitive to most inhibitors tested. The enzyme lost over 80 % of activity in the presence of 2 mM of PMSF, indicating it is a serine protease (Table 4) (Powers et al. 2002). Furthermore, the activity was enhanced in the presence of the reducing agent DTT, showing 183 % of initial activity after pre-incubation with 5 mM of the agent. The presence of 2 mM EDTA reduced activity to 20 % of the control, suggesting that metal cations play an essential role in enzyme activation and/or stabilization (Table 4). Inhibition of the protease in the presence of EDTA can limit its use in laundry detergents which typically contain chelators, but offers it potential for use in applications carried out in hard water or under conditions where removal of cations is time consuming and not cost effective.
Effect of metal ions on protease activity
It has been reported that the majority of halophilic, alkaliphilic serine proteases require calcium and magnesium for activity (Maruthiah et al. 2013). The protease from Alkalibacillus sp. NM-Da2 was active in the presence of 2 mM of CaCl2, MgCl2 and FeCl2, but activity was inhibited in the presence of 5 mM of each salt (Table 5). The protease lost approximately half of its initial activity in the presence of 2 mM of BaCl2, MnCl2 and CuCl2. No activity was exhibited in the presence of HgCl2, ZnSO4, SrCl2, AgNO3, CsCl2 and NH4Cl (both 2 and 5 mM). Stability of halo-alkaliphilic proteases in the presence of Ca2+ and Mg2+ has been reported (Maruthiah et al. 2013, 2015).
Bioremediation application
Protease was able to degrade dissolved proteins in a synthetic wastewater system. Fifty percent degradation was achieved after 5 h of incubation at 55 °C and pH 9 in the presence of 2.6 M NaCl (Fig. 7). The enzyme concentration achieving maximal degradation was 2 % (w/v), increasing the enzyme concentration resulted in decreased degradation efficiency. This could be due to aggregation of excess enzyme.

Application of protease in bioremediation. Protein removal in synthetic wastewater by protease added at different concentrations, 1 % (filled triangle), 2 % (filled square) and 3 % (w/v)(unfilled circle) under standard assay conditions
Many industries, such as leather, agricultural and food industries generate hypersaline wastewater. Such wastewater generally is alkaline and contains a high amount of organic content (Lefebvre and Moletta 2006). Release of this wastewater to the environment without pre-treatment adversely affects water quality and aquatic life. Hypersaline effluents are usually treated chemically, as biological treatments are inhibited by high salt concentration and alkaline pH (Lefebvre and Moletta 2006). Activity of protease from Alkalibacillus sp. NM-Da2 gives it great potential to be used as part of an enzyme cocktail that provides an environmentally friendly alternative for treatment of hypersaline wastewater.
Conclusions
An extremely alkaliphilic, halophilic, thermostable and oxidizing agent stable protease was produced by Alkalibacillus sp. NM-Da2, isolated from the soda lakes of the Wadi An Natrun, Egypt. Properties distinguishing this protease from other reported serine proteases include maximal proteolytic activity at combined extremes of 2.7–2.9 M NaCl, pH55 °C 9–10 and 54–58 °C and activity in the presence of oxidizing agents and organic solvents. The stability of the enzyme at high salt concentration as well as its ability to function in organic solvents make it an attractive candidate for catalyzing biosynthetic and esterification reactions which are commonly performed in nonaqueous conditions. Activity of the enzyme at alkaline pH values also gives it potential for application in bioremediation of polluted, hypersaline waste water.
References
Anson ML (1938) Estimation of cathepsin and the partial purification of cathepsin. J Gen Physiol 22:79–89
Arabaci N, Yasemin C, Maasoglu Y, Arikan B (2013) Partial purification and characterization of thermostable, alkaline and chelator-resistant protease from a newly isolated Bacillus sp. CY7 and its potential applications in various industries JABS 7:14-19
Borkar S (2015) Alkaliphilic bacteria: diversity, physiology and industrial applications. In: Borkar S (ed) Bioprospects of Coastal Eubacteria. Springer International Publishing, Switzerland, pp 59–83
Bradford MM (1976) A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chanalia P, Gandhi D, Jodha D, Singh J (2011) Applications of microbial proteases in pharmaceutical industry: an overview Rev Med Microbiol 22:96–101
DasSarma S, DasSarma P (2015) Halophiles and their enzymes: negativity put to good use. Curr Opin Microbiol 25:120–126
Delgado-Garcia M, Valdvia-Urdiales B, Aguilar-Gonzalez CN, Contreras-Esquivel JC, Rodriquez-Herrera R (2012) Halophilic hydrolases as a new tool for biotechnological industries. J Sci Food Agric 92:2575–2580
Gupta A, Khare SK (2009) Enzymes from solvent-tolerant microbes: useful biocatalysts for nonaqueous enzymology. Crit Rev Biotechnol 29:44–54
Gupta R, Beg QK, Lorenz P (2002) Bacterial alkaline proteases: molecular approaches and industrial applications. Appl Microbiol Biotechnol 59:15–32
Jain D, Pancha I, Mishra SK, Shrivastav A, Mishra S (2012) Purification and characterization of haloalkaline thermoactive, solvent stable and SDS-induced protease from Bacillus sp: a potential additive for laundry detergents. Bioresour Technol 115:228–236
Jisha VN et al (2013) Versatility of microbial proteases. Adv Enzyme Res 1:39–51
Joo HS, Kumar CG, Park GC, Palik SR, Chang CS (2003) Oxidant and SDS-stable alkaline protease from Bacillus clausii I-52: production and some properties. J Appl Microbiol 5:267–272
Joshi S, Satyanarayana T (2013) Characteristics and applications of a recombinant alkaline serine protease from a novel bacterium Bacillus lehensis. Bioresour Technol 131:76–85
Karbalaei-Heidari HR, Shahbazi M, Absalan G (2013) Characterization of a novel organic solvent tolerant protease from a moderately halophilic bacterium and its behavior in ionic liquids. Appl Biochem Biotechnol 170:573–586
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lee M, Dordick JS (2002) Enzyme activation for nonaqueous media. Curr Opin Biotechnol 13:376–384
Lefebvre O, Moletta R (2006) Treatment of organic pollution in industrial saline wastewater: a literature review. Water Res 40:3671–3682
Maruthiah T, Esakkiraj P, Prabakaran G, Palavesam A, Immanuel G (2013) Purification and characterization of moderately halophilic alkaline serine protease from marine Bacillus subtilis AP-MSU 6 Biocat Agri. Biotechnol 2:116–119
Maruthiah T, Immanuel G, Palavesam A (2015) Purification and characterization of halophilic organic solvent tolerant protease from marine Bacillus sp. APCMST-RS7 and its antioxidant potentials Proc Natl Acad Sci, India, Sect B Biol Sci. doi:10.1007/s40011-40015-40603-40010
Mesbah NM, Wiegel J (2012) Life under multiple extreme conditions: diversity and physiology of the halophilic alkalithermophiles App. Environ Microbiol 78:4074–4082
Mesbah NM, Wiegel J (2014) Purification and biochemical characterization of halophilic, alkalithermophilic protease AbCP from Alkalibacillus sp. NM-Fa4. J Mol Catal B Enzymatic 105:74–81
Ogino H, Ishikawa H (2001) Enzymes which are stable in the presence of organic solvents. J Biosci Bioeng 91:109–116
Oren A (2010) Industrial and environmental applications of halophilic microorganisms. Environ Technol 31:825–834
Powers JC, Asgian JL, Ekici OD, James KE (2002) Irreversible inhibitors of serine, cysteine and threonine proteases. Chem Rev 102:4639–4750
Raddadi N, Cherif A, Daffonchio D, Neifar M, Fava F (2015) Biotechnological applications of extremophiles, extremozymes and extremolytes. Appl Microbiol Biotechnol 99:7907–7913
Rosenberg L et al (2004) Safety and efficacy of a proteolytic enzyme for enzymatic burn debridement: A preliminary report. Burns 8:843–850
Sarethy IP, Saxena Y, Kapoor A, Sharma M, Sharma SK, Gupta V, Gupta S (2011) Alkaliphilic bacteria: applications in industrial biotechnology. J Ind Microbiol Biotechnol 38:769–790
Sarmiento F, Peralta R, Blamey JM (2015) Cold and hot extremozymes: industrial relevance and current trends Front Bioeng. Biotechnol 3:148
Schmidt TM, Bleakley B, Nealson KH (1988) Characterization of an extracellular protease from the insect pathogen Xenorhabdus luminescens App. Environ Microbiol 54:2793–2797
Selim S, Hagagy N, Abdel-Aziz M, El-Meleigy E, Pessione E (2014) Thermostable alkaline halophilic-protease production by Natronolimnobius innermongolicus WN18. Nat Prod Res 28:1476–1479
Singh SK, Singh SP, Tripathi VR, Garg SK (2012) Purification, characterization and secondary structure elucidation of a detergent stable, halotolerant, thermoalkaline protease from Bacillus cereus SIU1. Process Biochem 47:1479–1487
Sinha R, Khare SK (2013) Characterization of detergent compatible protease of a halophilic Bacillus sp. EMB9: Differential role of metal ions in stability and activity. Bioresour Technol 145:357–361
Synowiecki J (2015) Some applications of thermophiles and their enzymes for protein processing. Afr J Biotechnol 9:7020–7025
Wiegel J (1998) Anaerobic alkalithermophiles, a novel group of extremophiles. Extremophiles 2:247–267
Wilson K (1997) Preparation of genomic DNA from Bacteria. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (eds) Current Protocols in Molecular Biology. Wiley, New York, pp 2.4.1–2.4.5
Yin J, Chen J-C, Wu Q, Chen G-Q (2015) Halophiles, coming stars for industrial biotechnology. Biotechnol Adv 33:1433–1442
Acknowledgments
This work was supported by the US, Egypt Science and Technology Joint Fund in cooperation with the Suez Canal University (Egypt) under Project number 1841 and the University of Georgia (USA) under Project Number NSF-OISE-1132412.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by L. Huang.
Rights and permissions
About this article

Cite this article
Abdel-Hamed, A.R., Abo-Elmatty, D.M., Wiegel, J. et al. Biochemical characterization of a halophilic, alkalithermophilic protease from Alkalibacillus sp. NM-Da2. Extremophiles 20, 885–894 (2016). https://doi.org/10.1007/s00792-016-0879-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-016-0879-x