
Abstract
In Archaea, type IV prepilins and prearchaellins are processed by designated signal peptidase III (SPaseIII) prior to their incorporation into pili and the archaellum, respectively. These peptidases belong to the family of integral membrane aspartic acid proteases that contain two essential aspartate residues of which the second aspartate is located in a conserved GxGD motif. To this group also bacterial type IV prepilin peptidases, Alzheimer disease-related secretases, signal peptide peptidases and signal peptide peptidase-like proteases in humans belong. Here we have performed detailed in vivo analyses to understand the cleavage activity of PibD, SPaseIII from the thermoacidophilic crenarchaeon Sulfolobus acidocaldarius. Using an already established in vivo heterologous system cleavage assay, we could successfully identify the key amino acid residues essential for catalysis of PibD. Furthermore, in trans complementation of a pibD S. acidocaldarius deletion mutant with PibD variants having substituted key amino acids has consolidated our observations of the importance of these residues in catalysis. Based on our data, we propose to re-define class III peptidases/type IV prepilin/prearchaellin peptidases as GxHyD group (rather than GxGD) of proteases [Hy-hydrophobic amino acid].
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The pilins and archaellins that are forming the helical fiber of the type IV pili in archaea and bacteria are synthesized with a signal peptide III at the N-terminus of the preprotein (Pohlschroder et al. 2011; Szabo et al. 2007) that includes a short sequence of positively charged amino acids followed by a signal peptidase cleavage site. In contrast to signal peptide I and II (class I and class II signal peptides), the hydrophobic stretch in signal peptide III (class III signal peptide) is located after the cleavage site and it facilitates the anchoring of processed pilins and archaellins to the membrane. Prior to incorporation into the filament, this signal peptide III is processed by a dedicated class III peptidase (SPaseIII) (Craig and Li 2008; Hansen and Forest 2006), enabling assembly of the mature pilins via the hydrophobic alpha helix at the newly formed N-terminus. The SPaseIII found in bacteria and archaea belongs to the family of integral membrane aspartic acid proteases that possess two essential aspartic acid residues of which the second one is embedded in a GxGD motif (LaPointe and Taylor, 2000). Moreover, these aspartates are located within two transmembrane domains of the peptidase but will be brought to the cytoplasm due to conformational changes of the enzyme during cleavage activity (Hu et al. 2011). Next to cleavage activity, the bacterial SPasesIII were found to transfer a methyl group to a phenylalanine residue at the processed N-terminus of the mature type IV pilin. However, the catalytic centre for cleavage activity and methylation are separated (Strom et al. 1993) and in contrast to cleavage, methylation of the pilin is not a prerequisite for the subsequent incorporation of the pilins into the growing fiber (Pepe and Lory 1998).
In Archaea, SPasesIII from the crenarchaeon Sulfolobus solfataricus (PibD) (Albers et al. 2003; Szabo et al. 2006; Szabo et al. 2007) and euryarchaea Methanococcus maripaludis, Methanococcus voltae (FlaK) (Bardy and Jarrell 2002, 2003; Ng et al. 2009; Hu et al. 2011) and Haloferax volcanii (Tripepi et al. 2010) have been characterized in vitro and in vivo. Unlike bacteria, archaeal SPasesIII do not possess methyltransferase activity, however it was shown for PibD in S. solfataricus and FlaK in M. maripaludis that two conserved aspartarte residues are essential for substrate processing as well (LaPointe and Taylor, 2000, de Bentzmann et al. 2006, Szabo et al. 2006, Bardy and Jarrell, 2003). In 2011, the crystal structure of FlaK from M. maripaludis was solved bearing six transmembrane helices with the first essential aspartic acid located in helix 1 and the second in the conserved GxGD motif in helix 4 that will be brought to close proximity for catalysis by undergoing conformational changes (Hu et al., 2011). Additionally, structural comparison of the FlaK crystal with a model of presenilin, a GxGD protease involved in processing of amyloid precursor proteins (APP) in humans (Wolfe 2013), has further led to the conclusion that prokaryotic and human proteases are evolutionary related (Hu et al. 2011).
Contrary to FlaK, which is a prearchaellin peptidase and therefore exclusively processes prearchaellins, the subunits forming the archaellum, PibD from S. solfataricus and H. volcanii have been reported to have a broad substrate specificity including both prearchaellins and type IV prepilins (Albers et al. 2003; Tripepi et al. 2010). Studies on PibD from S. solfataricus have revealed that the substrate range of PibD includes prearchaellins, UV-induced type IV pilin-like proteins (ups) and the sugar binding protein of the bindosome assembly system (BasA/BasB/BasC) (Albers et al. 2003; Frols et al. 2008). However, it is not clear how PibD from the Sulfolobales has been naturally engineered and selected to process a broad range of substrates involved in different physiological processes in contrast to other archaea that seem to have dedicated SPasesIII for different kinds of substrates. In M. maripaludis, an additional SPaseIII, EppA, has been described (Szabo et al. 2007) which only processes prepilins that contain a DUF361 domain.
Interestingly, a deletion of PibD in S. solfataricus was found to be non-viable showing its physiological indispensability in this organism (Albers and Pohlschroder 2009; Lassak et al. 2012; Pohlschroder et al. 2011). An in vitro aspartyl-mutational scan of PibD from S. solfataricus revealed that aspartyl residues D23 and D80 are essential for catalysis. Additionally, residues D187 and D188 were found to be important for processing (Szabo et al. 2006). However, the lack of structural insights on class III peptidases/type IV pilin peptidases so far limited our understanding on the catalytic nature of PibD.
In this study, we have performed detailed in vivo analyses to shed light on the catalytic potential of PibD from Sulfolobus acidocaldarius. In addition to heterologous cleavage assays in E.coli (Szabo et al. 2007; Henche et al. 2012), we used in vivo cleavage assays in S. acidocaldarius, that allowed us to monitor the catalytic activity of PibD and several PibD single mutants. Thereby we could identify residues in PibD important for substrate processing. Based on our data we propose to re-define SPaseIII as GxHyD group (rather than GxGD) of protease [X-any amino acid, Hy-hydrophobic amino acid].
Materials and methods
Strains and growth conditions
S. acidocaldarius DSM639 was grown aerobically at 75 °C in Brock’s basal salts medium (Brock et al. 1972) adjusted to pH 3.5 with sulphuric acid and supplemented with 0.1 % (w/v) tryptone (Roth) or N-Z-amine AS (Sigma) and 0.2 % (w/v) dextrin. For the uracil auxotrophic strains, S. acidocaldarius MW001 (containing a 322 bp deletion in the pyrE gene) (Wagner et al. 2012), S. acidocaldarius pibD mutant strains (MW114, ΔpibD in S. acidocaldarius MW001 background, (see Table 1), the growth medium was supplemented with 10 µg ml−1 uracil. To prepare plates, Brock medium was solidified by adding a final concentration of 0.6 % (w/v) gelrite (Sigma), 10 mM MgCl2 and 3 mM CaCl2. Plates were then incubated for 5 days at 75 °C. For the propagation of plasmids E. coli strain DH5α was used. For the methylation of the plasmids E.coli ER1821kan containing pM.EsaBC4I (New England Biolabs) was used. Heterologous expression analysis was performed in E.coli BL21(DE3) containing a RIL plasmid that encodes additional tRNAs for rare codons (Stratagene).
Cloning and plasmid construction
To study the in vivo cleavage of the prepilin AapA and prearchaellin FlaB2 by peptidases PibD and FlaK, a co-expression vector was constructed harboring the signal peptide III (SPaseIII) containing substrates and the archaeal prepilin peptidases under differentially regulated promoters (Henche et al. 2012). Therefore, S. acidocaldarius aapA (saci_2314) and M. maripaludis flaB2 (MMP_1667) were amplified and cloned into pZA7 (Szabo et al. 2006) using restriction enzymes NcoI and BamHI, which adds a C-terminal hemagglutinin (HA)-tag to the introduced gene. Subsequently, the C-terminally tagged genes were cloned into pBAD/Myc-HisA (Invitrogen) via NcoI and HindIII resulting in pSVA906 (aapA + HA) and pSVA2328 (flaB2 + HA) with an L-arabinose-inducible promoter upstream of the gene of interest (Table S1). For co-expression of these proteins with SPaseIII PibD or FlaK, the genes pibD (saci_0139) and flaK (MMP_0555) were first cloned into the vector pUC18-pibD as described previously (Szabo et al. 2007), by adding a T7 promoter and a C-terminal six-histidine tag to the genes. These cassettes were then cloned into the plasmids pSVA906 and pSVA2328 using the restriction enzyme SphI, yielding in pSVA914 (aapA + HA, pibD + 6xHis), pSVA2313 (aapA + HA, flaK + 6xHis), pSVA2315 (flaB2 + HA, pibD + 6xHis) and pSVA2329 (flaB2 + HA, flaK + 6xHis) (Table S1). For the study of single amino acid mutations in the peptidases, the vectors pSVA914, pSVA2313, pSVA2315 and pSVA2329 were amplified using primers containing the desired amino acid substitution in PibD or FlaK (Table S2). Subsequently, the amplified vectors were treated with the restriction enzyme DpnI in order to digest all methylated plasmids. Thus, only newly synthesized non-methylated plasmids were transformed into E. coli DH5α cells. Plasmids were isolated and checked by sequencing for the correct single amino acid substitution.
The plasmids used for the in trans complementation of pibD were constructed by amplifying the pibD gene from S. acidocaldarius MW001 genomic DNA with primers listed in Table S2. The PCR product was cloned into pMZ1 using restriction enzymes NcoI and BamHI, yielding plasmid pSVA381 in which the gene pibD was C-terminally tagged with a 6xHis/Strep-tag. The respective single amino acid substitutions were created by amplifying the plasmids with primers that contained the preferred mutation, as described above. Next, the sequenced plasmids were digested with NcoI and EagI and the inserts were cloned into pSVA1450, in which the C-terminally-tagged pibD was under the control of a maltose-inducible promoter. Before electroporation into Sulfolobus cells, correct plasmids were methylated in E. coli ER1821 containing the additional plasmid pM.EsaBC4I (New England Biolabs).
Expression of recombinant genes in E.coli and in vivo cleavage assay
The above described plasmids (Table S1) were transformed into E. coli BL21 (DE3) RIL cells (Agilent Technologies). Expression of the pili genes was induced by the addition of 0.2 % L-arabinose, and PibD expression was initiated by the addition of 0.25 mM IPTG. After 4 h of induction, the cells were centrifuged, resuspended in 4 ml of Hepes buffer (50 mM Hepes pH 7.5, 500 mM NaCl, 10 % glycerol, 1X protease inhibitor cocktail (Roche) and lysed by sonication with a Sonoplus HD3100 sonicator (Bandelin Sonorex Biotechnique, Germany) using microtip MS 73. After a low spin (4500g, 20 min) and an ultracentrifugation step (250, 000g, 45 min) the membrane fraction was resuspended in 100 µl of 50 mM Hepes pH 7.5, 500 mM NaCl, 10 % glycerol, 1X protease inhibitor cocktail. The samples were analyzed by SDS-PAGE and AapA was immunodetected using monoclonal anti-hemagglutinin antibody (Sigma-Aldrich).
As the expression level of PibD was too low to be directly visualized by Western immunoblotting, an enrichment step was introduced for each of the samples. Membranes in amounts corresponding to two times the amount used in each cleavage reaction were solubilized by the addition of 1 % (vol/vol) Triton X-100 in a total volume of 0.5 ml. The mixture was incubated for 1 h at 37 °C and centrifuged in a TLA100.2 rotor for 20 min at 220,000 × g and 4 °C. Under these conditions, PibD was found exclusively in the pellet fraction (not shown). The pellet was resuspended in 40 µl sodium dodecyl sulfate (SDS) sample buffer, and proteins were separated on a 12 % SDS-polyacrylamide gel electrophoresis (PAGE) gel and blotted onto a polyvinylidene difluoride membrane (Roche, Mannheim, Germany). The C-terminal six-histidine tag on PibD was detected using an anti-His tag antibody (Abcam).
Generation of in frame deletion of pibD
The markerless deletion of the pibD gene in the uracil-auxotrophic S. acidocaldarius strain MW001 was generated using the classical “pop in/pop out” method described by Wagner et al. (2012) which is based on two consecutive homologous recombination events. In short, around 1 kb of the respective up-and downstream flanking regions of pibD were amplified by PCR using the corresponding primers listed in Table S2 and S. acidocaldarius genomic DNA as a template. By overlap extension PCR, the up- and downstream flanking regions were connected and amplified using the outward bound primers. The PCR product was subsequently cloned into pSVA406 that contains the genes pyrEF, resulting in pSVA1806 (see Table S1). The plasmid was methylated in E. coli ER1821 and transformed into MW001. Integrants were selected on plates lacking uracil and grown in 24-well plates for 2 days in liquid first selection medium. Next, the cultures were plated and grown for 5 days on second selection plates containing uracil and 100 µg/ml 5-FOA. Under these conditions, the plasmid is forced to loop out by homologous recombination. Obtained colonies were tested by PCR for a successful deletion of the gene. Deletion of pibD was confirmed by DNA sequencing resulting in strain MW114.
In trans complementation of pibD deletion mutants
Recombinant plasmids were constructed using standard molecular biology techniques described under the ‘cloning and expression’ and ‘Construction of plasmids for gene deletion’ sections and are listed in Table 1. PibD was cloned under the control of the maltose-inducible promoter malA into pSVA1450, a pCMalLacS derived expression plasmid (Berkner et al. 2007). To complement the ΔpibD deletion strain, constructs containing wild type and mutated pibD variants (Table S1) were electroporated in electrocompetent ΔpibD deletion mutant cells. After electroporation, cells were grown in Brock medium containing 0.1 % N-Z-amine and 0.1–0.4 % maltose. The expression of the target genes was verified by SDS-PAGE followed by Western blotting using α-His antibodies.
Electron microscopy
The different S. acidocaldarius strains were grown to an OD600nm 0.8 and fixed with 1.25 % glutaraldehyde. After 15 min, the cells were collected and applied to hydrophobilized carbon-coated nickel- or copper grids, washed twice with water and negatively stained with 2 % uranyl acetate and air-dried. Images were obtained on a JEOL JEM-2100 transmission electron microscope (JEOL, Japan) at an acceleration voltage of 120 kV and with a 2k × 2k fast scan CCD camera F214 (TVIPS, Germany).
Results
Cloning and expression of PibD from S. acidocaldarius
To identify the type IV prepilin-like signal peptidase, we first searched the S. acidocaldarius genome database (The Sulfolobus database: MUTAGEN; http://www.sulfolobus.org/) for a putative homolog of PibD (Sso_0131) of S. solfataricus. Thereby, we identified a putative PibD homolog: saci_0139, although the annotated gene fragment was found significantly shorter [92 amino acids (aa) shorter] than the full-length S. solfataricus PibD (236 aa for Sso_PibD). As this annotation seemed to be incorrect, we performed further in silico analysis of the transmembrane topology and based on our analysis we assigned three putative open reading frames (ORF-1, 234 aa; ORF-2, 222 aa; and ORF-3, 186 aa until the stop codon) for further analysis (Fig. 1a). All three possible open reading frames for the putative pibD gene were cloned into the S.acidocaldarius expression vector [pSVA381 (ORF-1), pSVA382 (ORF-2) and pSVA383 (ORF-3)] and used for complementation of ΔpibD mutant of S. acidocaldarius MW001. The deletion strain ΔpibD did not exhibit any type IV pilus-like structures when analyzed by transmission electron microscopy showing that the assembly of archaellum, Ups and Aap pili is dependent on the SPaseIII PibD in S. acidocaldarius (Fig. 4). Cell aggregation and attachment to surfaces was strongly reduced in the pibD deletion strain and no growth defect was observed (data not shown). Electron microscopic analysis and cell-aggregation assays confirmed that construct pSVA381 (ORF-1) could complement the pibD deletion phenotype (Fig. 4, data not shown). Subsequent experiments were therefore performed using the newly assigned open reading frame (ORF-1) for the pibD gene (saci_0139).

a Amino acid sequence of S. acidocaldarius PibD. The start points of the different tested version of PibD (ORF-1, ORF-2, ORF-3) are indicated by arrows. Mutated amino acids are underlined. b Predicted membrane topology of PibD from S. acidocaldarius. Different amino acids that were mutated in this study are marked in the membrane topology model. The residues forming the catalytic center of PibD are in red circles, residues that are conserved in Sulfolobales are in green circles and residues that were reported to influence the catalytic activity of FlaK in M. maripaludis are in yellow circles
Wild-type PibD and different point mutants were employed to complement the ΔpibD mutant in S.acidocaldarius MW001 (MW114). Trans-complementation with wild-type PibD could restore the wild-type phenotype in a ΔpibD mutant, as surface structures could again be observed (Aap pili, Ups pili and archaella). However, certain point mutants of pibD resulted in different level of phenotypic complementation as evident from electron microscopy and Western blot analyses (Figs. 2, 4).

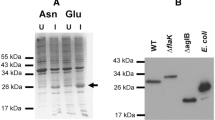
In vivo processing of AapA by PibD and PibD mutants in E.coli. The substrate AapA was detected by Western blot analysis using HA-specific antibodies. The full-length AapA has a size of 17.6 kDa. After processing by PibD, AapA displays band at 15 kDa. The expression of PibD was confirmed by immunoblot analysis using His-specific antibodies. PibD has a size of 28 kDa
For heterologous cleavage assays in E.coli, pre-AapA, which is the filament protein of the aap pilus, and was shown to be processed by the PibD in S. acidocaldarius, was selected as the substrate (Henche et al. 2012). An in vivo cleavage assay in E. coli was performed in which the expression of the pilins was induced by the addition of arabinose two hours prior to the induction of PibD by the addition of IPTG. Membrane extracts of the expressing E. coli strains were separated on SDS-PAGE and AapA was detected by immunoblotting using anti-HA antibodies. The full-length AapA has a size of 17.6 kDa, whereas the processed AapA is around 15 kDA. Different point mutants of PibD showed differential activity allowing an identification of the residues key to the catalysis (see next paragraph).
Identification of catalytic residues in PibD by site-directed mutagenesis
To determine which residues in PibD are essential for catalysis, a recent crystal structure of FlaK from M. maripaludis was employed to build a model of PibD. Unfortunately, low sequence homology between FlaK and PibD as well as the presence of multiple membrane spanning domains resulted in a PibD structural model with low confidence (Fig. 1b). With structure based local sequence alignment however, we were able to identify possible amino acid residues involved in catalysis in PibD (Fig. 1). Using available information from previous analyses of Sso_PibD (Sso_0131) and identified catalytic residues in FlaK (MMP_0555), we constructed five active site mutants representing the GxGD motif. The catalytic aspartate residues (D21 and D78), glycine residues (G75 & G76) and alanine residue (A77) were changed to alanine or glutamate by site-directed mutagenesis. In addition, amino acid residues that are conserved in PibD from all Sulfolobus spp. (L81, I22, K23,K24) and amino acid residues which were shown to influence the catalytic activity of FlaK (R25, W32, L79, P215), were mutated either to alanine or glutamate. In a previous study, D185 and D186 were shown to be important in PibD of S. solfataricus (position in S. solfataricus PibD: D187 and D188) (Szabo et al. 2006). To further evaluate the roles of these residues, D185 and D186 were changed into alanine by site-directed mutagenesis. All these mutant proteins were expressed in E.coli. Western blot analysis confirmed PibD/FlaK expression and revealed equal amounts of expression in all mutants (Fig. 2, 3, lower panels). The same membrane fractions were used for detection of cleavage of the type IV pre-pilin-like signal peptide on pre-AapA using anti-HA antibodies.

In vivo processing of AapA/FlaB2 by PibD/FlaK and PibD/FlaK in E.coli. The substrate AapA was detected by Western blot analysis using HA-specific antibodies. The full-length AapA has a size of 17.6 kDa. After processing by PibD, AapA displays band at 15 kDa. The size of the full-length FlaB2 is around 22.5 kDa, whereas the processed FlaB2 has a size of 20.9 kDa. The expression of PibD or FlaK was checked by immunoblot analysis using His-specific antibodies. PibD has a size of 28 kDa whereas FlaK has size of 25 kDa
Unlike FlaK, where GxGD forms the catalytic region, PibD was found to contain a GxAD motive. Analysis of the single mutation of the catalytic residues G75E, A77E and D78A revealed no processing of the pre-AapA confirming that these residues are indeed forming the catalytic core in S. acidocaldarius PibD (Fig. 2a). In PibD, a glycine residue is part of the GxAD motif, representing the amino acid X, which was mutated to a glutamic acid residue. This mutation did not abolish cleavage activity of PibD (Fig. 2a), confirming that this position is not essential for catalytic activity. The other catalytic important residue is D21, which is located in the first membrane domain of FlaK and PibD. Also, this residue was identified to be essential as the D21A mutant did not show any activity towards the substrate.
Previously, it was shown for S. solfataricus PibD that aspartate D185 and D186 are essential for catalysis as the mutation of these residues resulted in a reduction of the activity (Szabo et al. 2006). In FlaK however, structural and biochemical analyses have shown that these two residues are not essential for catalysis (Hu et al. 2011). In PibD from S. acidocaldarius, we also observed normal activity of D186A and a lower activity for D185A mutant (Fig. 2b). The activity levels of single mutants, I22E, K23A, K24A, L81A and D186A, residues that are conserved among all Sulfolobales PibD enzymes, were comparable to that of the wild type enzyme under standard assay conditions (Fig. 2b). Thus, these residues do not seem to be important for substrate processing.
Similar to what was observed for FlaK, single mutants R25A, L79A and P215A were found essential for catalysis in Saci_PibD. However, the mutation of W32A only showed a reduced activity of PibD, indicating that this residue is not essential to cleave the substrate in contrast to FlaK (Fig. 2a).
As was described before, PibD harbors a GxAD motive instead of a GxGD motive. Given the fact that the exchange of glycine to an alanine did not abolish cleavage activity, but the exchange to glutamic acid did, we speculate that it is important to have a non-charged amino acid at this position. To confirm this assumption, PibD mutants were created that harbor different amino acid substitutions at position A77 which were A77G, A77P and A77K (Fig. 3a). Analysis of these mutants revealed processing of the the pre-AapA substrate when the position was exchanged with a non-polar amino acid (A77G and A77P), although the cleavage activity for A77P was reduced in comparison to the wild type. No catalytic activity was observed when A77 was changed to a charged polar amino acid such as glutamic acid or lysine (A77E and A77K), confirming the hypothesis that a GXHyD motif is important for cleavage (Fig. 3a). In contrast to PibD that can cleave different signal peptide III containing proteins found in S. acidocaldarius, FlaK from M. maripaludis was reported to only process the pre-archaellin FlaB2 and not the DUF361 domain containing prepilins EpdA and EpdC (Szabo et al. 2007) (Fig 3b). To test whether this specificity is caused by the difference in the GxHyD motif, FlaK and a G78A mutant of FlaK were tested with AapA as a substrate; no activity could be observed for either the wild type or the mutated FlaK (Fig. 3a). Given the fact that FlaK shows no activity towards AapA as a substrate, we could not test if the modified GxAD motive is also functional in FlaK. Therefore we constructed plasmids that contain the FlaK-specific substrate FlaB2 (MMP1667) from M. maripaludis. Using this substrate, we were able to show that both wild-type FlaK and the G78A mutant were catalytically active (Fig. 3b). FlaK conclusively also requires a GxHyD motif rather than a GxGD motive. Surprisingly, only the PibD A77G mutant is able to process the substrate FlaB2 but not the PibD with the wild-type GxAD motif (Fig. 3b).
To summarize, here we identified key residues in PibD from S. acidocaldarius which are essential for catalysis. Notably, we identified a GXAD motif instead of the known GxGD in the catalytic centre of Saci_PibD. We speculate that a GXHyD-motif is necessary and sufficient for the catalytic activity of SPaseIII as the mutation to polar amino acids like glutamic acid and lysine (mutations A77E and A77K) abolished cleavage activity, whereas the substitution a to non-polar amino acids (mutations A77G and A77P) did not eliminate substrate processing.
To further evaluate the catalytic potential of PibD, wild type and single mutants were expressed in trans in a ΔpibD mutant of S. acidocaldarius using a maltose-inducible plasmid. Cells harboring the respective plasmids were analyzed by transmission electron microscopy in order to check for the presence of Aap pili and thus for the catalytic activity of the PibD single mutants. The pibD deletion could be complemented with the wild-type PibD as numerous Aap pili could be detected on the cells (Fig. 4). When the deletion was complemented with mutants harboring mutations in the catalytic center (D21A, G75E, A77E and D78A), no Aap pili were detected, confirming that these residues for are also essential for PibD enzyme activity in S. acidocaldarius. When complemented with the G76E mutant, which is located within the GxGD motif, Aap pili were visible on the cells, verifying the result from the E. coli cleavage assay. All other PibD single mutants were analyzed by electron microscopy and gave results that were as predicted based on the cleavage assays (Figure S1). However, the R25A single mutant displayed Aap pili even though no AapA-processing was observed in the E. coli cleavage assays (Fig. 2a).

Electron micrographs showing the S. acidocaldarius pibD deletion mutant in (a) and the pibD deletion mutant complemented with different plasmids expressing the indicated pibD mutants. The cells were negatively stained with uranyl acetate. All surface structures visible were aap pili. Scale bars are 200 nm
Discussion
The SPaseIII belong to the family of aspartate proteases. However, in contrast to the classical aspartyl proteases, which harbor a D(T/S)G(T/S) active site motif (Pearl and Blundell 1984), the bacterial and archaeal prepilin/prearchaellin peptidases contain two essential aspartyl residues of which the C-terminal aspartate is located in a conserved GxGD motif (LaPointe and Taylor 2000). This family of integral membrane aspartic acid proteases comprises Alzheimer disease-related secretases, signal peptide peptidases (Wolfe 2010) and signal peptide peptidase-like proteases in humans (Voss et al. 2013; Tomita and Iwatsubo 2013) which share the property of the conserved GxGD motif. Though, this group of eukaryotic proteases belongs to the intra-membrane cleaving proteases (I-CLiPs), as their essential aspartyl residues are localized within the transmembrane domains and the cleavage of the substrates occurs inside the membrane (Golde et al. 2009; Fluhrer et al. 2009). In contrast to this, the catalytically important aspartate residues in the prokaryotic peptidases are located near the membrane-cytosol interface. Nevertheless, the residues are still part of the transmembrane domain but will be brought into the cytoplasm due to conformational changes in the enzyme (Hu et al. 2011).
In the present work, we have shown that SPaseIII PibD from S. acidocaldarius shares the signature of two conserved active site motifs comprising of key aspartyl residues. However, contrary to the described conserved GxGD motif, we identified a GxAD motif in PibD from S. acidocaldarius. Moreover, an in silico analysis additionally revealed the presence of an altered GxGD motif in many other SPaseIII. In fact, about 60 % of all analyzed archaeal peptidases (Figure S3) harbored a GxAD motif. This motif was predominantly found in halophilic archaea, most of the crenarchaeal and in all kor- and thaumarchaeal species. However, for methanogenic archaea both motifs seemed to be equally distributed. As either a glycine or an alanine residue was identified at position +3 in the GxHyD motif, it is very likely that the SPaseIII has its best enzymatic activity when a small non-polar amino acid is present at this position. Nevertheless, cleavage was still observed when the alanine was mutated to a bigger hydrophobic amino acid like proline. At position +2 in the GxHyD motif four different amino acids were identified: glycine, serine, leucine and glutamic acid. Interestingly, the two SPaseIIIs from Methanococcus voltae contained different motifs, the FlaK homolog possesses the GxAD motif whereas the EppA homolog harbors the GxGD motif. In contrast to this, both SPaseIII in M. maripaludis contain the GxGD motif. Therefore, no direct correlation can be concluded for the presence of the GxHyD motif and different types of SPaseIIIs in archaea (prearchaellin or prepilin peptidase).
Combining our results and analysis, we therefore hypothesized that a GxHyD-motif at the active site is sufficient for the catalytic activity of type IV prepilin-like peptidases. Site-directed mutagenesis of D21 and D78, the two essential aspartate residues, resulted in inactivation of the PibD catalytic activity confirming the previous observation of their importance in catalysis. Further attempts to perform detailed mutagenesis of the GxAD motif revealed that whenever the alanine (A77) residue was mutated to a nonpolar amino acid, PibD was still catalytically active, although the cleavage activity was reduced in case of the A77P mutant. Similarly, an exchange of the M. maripaludis FlaK G78 to alanine in the GxGD motif also resulted in a cleavage activity comparable to the wild-type enzyme, confirming our hypothesis of the GxHyD active site motif. However, when PibD was tested for its ability to cleave FlaB2 from M. maripaludis, the enzyme was only able to process the prearchaellin when the GxAD was changed to the classical GxGD motif. We believe that this observation is indicative of the requirement for proper conformational topology that governs the recognition of specific substrates. However, when in FlaK the motif was mutated into the “relaxed” version, it could still not process the pre-AapA pilin, indicating that properties, other than this motif, are involved in the substrate specificity of these peptidases.
Site-directed mutagenesis of the PibD residues, which were shown to be indispensible for catalysis of FlaK, displayed a reduced catalytic activity confirming that the amino acids R25, L79 and P215 are also essential in PibD. Given the fact that of the latter residues, R25 and L79 lie in close proximity to the catalytically important aspartyl residues in Saci_PibD (D21 and D78) we hypothesize a role for R25 and L79 in coordinating the aspartate residues for proper cleavage. However, no cleavage was observed for the PibD_R25A mutant in E. coli. Conversely, Aap pili were formed when the respective mutation was analyzed in S. acidocaldarius. We therefore believe that this unexpected observation might be due to the different membranes and subsequent topology of PibD in E. coli and S. acidocaldarius. Moreover, our in vivo observations in S. acidocaldarius led us to propose that R25 is probably not involved in the catalysis.
Previously, aspartyl-scanning analysis of PibD in S. solfataricus revealed that next to the key catalytic aspartates, two other aspartate residues, located in a big cytoplasmic loop between TM5 and TM6, are also important for catalysis (Szabo et al. 2006). Moreover, it has been postulated that these residues might be involved in forming salt bridges and in substrate recognition by binding to the positively charged N-terminus of the prepilin. Contrary to this, for the PibD from S. acidocaldarius, we observed that alanine substitutions of D185 and D186 did not interfere with the cleavage, only a reduction of activity was observed for the D185A mutant. Similar results were also reported for FlaK; where E. coli cleavage assays and analysis of the FlaK crystal structure revealed that these residues are not essential for activity and not in the vicinity of the active site during catalysis (Hu et al. 2011). Site-directed mutagenesis of the amino acids that were conserved among the PibD homologs in Sulfolobales demonstrated that none of these residues, i.e., L81, I22, K23 and K24 were important for the processing of the prepilins; even though these amino acids are located close to the catalytic centers of the enzyme, a mutation to alanine did not change the cleavage properties of Saci_PibD.
In conclusion, we hereby re-define the GXGD motif containing SPaseIII as GXHyD motif containing peptidases and we could identify key amino acid residues that are involved in catalysis of both PibD from S. acidocaldarius and FlaK from M. maripaludis. With these results, we can now provide a convincingly uniform catalytic model that governs all SPaseIII from all three domains of life.
References
Albers SV, Pohlschroder M (2009) Diversity of archaeal type IV pilin-like structures. Extremophiles 13(3):403–410
Albers SV, Szabo Z, Driessen AJ (2003) Archaeal homolog of bacterial type IV prepilin signal peptidases with broad substrate specificity. J Bacteriol 185(13):3918–3925
Bardy SL, Jarrell KF (2002) FlaK of the archaeon Methanococcus maripaludis possesses preflagellin peptidase activity. FEMS Microbiol Lett 208(1):53–59
Bardy SL, Jarrell KF (2003) Cleavage of preflagellins by an aspartic acid signal peptidase is essential for flagellation in the archaeon Methanococcus voltae. Mol Microbiol 50(4):1339–1347
Berkner S, Grogan D, Albers SV, Lipps G (2007) Small multicopy, non-integrative shuttle vectors based on the plasmid pRN1 for Sulfolobus acidocaldarius and Sulfolobus solfataricus, model organisms of the (cren-)archaea. Nucleic Acids Res 35(12):e88
Brock TD, Brock KM, Belly RT, Weiss RL (1972) Sulfolobus: a new genus of sulfur-oxidizing bacteria living at low pH and high temperature. Arch Mikrobiol 84(1):54–68
Craig L, Li J (2008) Type IV pili: paradoxes in form and function. Curr Opin Struc Biol 18(2):267–277
de Bentzmann S, Aurouze M, Ball G, Filloux A (2006) FppA, a novel Pseudomonas aeruginosa prepilin peptidase involved in assembly of type IVb pili. J Bacteriol 188(13):4851–4860
Fluhrer R, Steiner H, Haass C (2009) Intramembrane proteolysis by signal peptide peptidases: a comparative discussion of GXGD-type aspartyl proteases. J Biol Chem 284(21):13975–13979
Frols S, Ajon M, Wagner M, Teichmann D, Zolghadr B, Folea M, Boekema EJ, Driessen AJ, Schleper C, Albers SV (2008) UV-inducible cellular aggregation of the hyperthermophilic archaeon Sulfolobus solfataricus is mediated by pili formation. Mol Microbiol 70(4):938–952
Golde TE, Wolfe MS, Greenbaum DC (2009) Signal peptide peptidases: a family of intramembrane-cleaving proteases that cleave type 2 transmembrane proteins. Sem Cell Dev Biol 20(2):225–230
Hansen JK, Forest KT (2006) Type IV pilin structures: insights on shared architecture, fiber assembly, receptor binding and type II secretion. J Mol Microbiol Biotech 11((3–5)):192–207
Henche AL, Ghosh A, Yu X, Jeske T, Egelman E, Albers SV (2012) Structure and function of the adhesive type IV pilus of Sulfolobus acidocaldarius. Environ Microbiol 14(12):3188–3202
Hu J, Xue Y, Lee S, Ha Y (2011) The crystal structure of GXGD membrane protease FlaK. Nature 475(7357):528–531
LaPointe CF, Taylor RK (2000) The type 4 prepilin peptidases comprise a novel family of aspartic acid proteases. J Biol Chem 275(2):1502–1510
Lassak K, Ghosh A, Albers SV (2012) Diversity, assembly and regulation of archaeal type IV pili-like and non-type-IV pili-like surface structures. Res Microbiol 163(9–10):630–644
Ng SY, VanDyke DJ, Chaban B, Wu J, Nosaka Y, Aizawa S, Jarrell KF (2009) Different minimal signal peptide lengths recognized by the archaeal prepilin-like peptidases FlaK and PibD. J Bacteriol 191(21):6732–6740
Pearl L, Blundell T (1984) The active site of aspartic proteinases. FEBS Lett 174(1):96–101
Pepe JC, Lory S (1998) Amino acid substitutions in PilD, a bifunctional enzyme of Pseusomonas aeruginosa. Effect on leader peptidase and N-methyltransferase activities in vitro and in vivo. J Biol Chem 273(30):19120–19129
Pohlschroder M, Ghosh A, Tripepi M, Albers SV (2011) Archaeal type IV pilus-like structures–evolutionarily conserved prokaryotic surface organelles. Curr Opin Microbiol 14(3):357–363
Strom MS, Bergman P, Lory S (1993) Identification of active-site cysteines in the conserved domain of PilD, the bifunctional type IV pilin leader peptidase/N-methyltransferase of Pseudomonas aeruginosa. J Biol Chem 268(21):15788–15794
Szabo Z, Albers SV, Driessen AJ (2006) Active-site residues in the type IV prepilin peptidase homologue PibD from the archaeon Sulfolobus solfataricus. J Bacterio 188(4):1437–1443
Szabo Z, Stahl AO, Albers SV, Kissinger JC, Driessen AJ, Pohlschroder M (2007) Identification of diverse archaeal proteins with class III signal peptides cleaved by distinct archaeal prepilin peptidases. J Bacteriol 189(3):772–778
Tomita T, Iwatsubo T (2013) Structural biology of presenilins and signal peptide peptidases. J Biol Chem 288(21):14673–14680
Tripepi M, Imam S, Pohlschroder M (2010) Haloferax volcanii flagella are required for motility but are not involved in PibD-dependent surface adhesion. J Bacteriol 192(12):3093–3102
Voss M, Schroder B, Fluhrer R (2013) Mechanism, specificity, and physiology of signal peptide peptidase (SPP) and SPP-like proteases. Biochim Biophysica Acta 1828(12):2828–2839
Wagner M, van Wolferen M, Wagner A, Lassak K, Meyer BH, Reimann J, Albers SV (2012) Versatile Genetic Tool Box for the Crenarchaeote Sulfolobus acidocaldarius. Frontiers Microbiology 3:214
Wolfe MS (2010) Structure, mechanism and inhibition of gamma-secretase and presenilin-like proteases. Biological chemistry 391(8):839–847
Wolfe MS (2013) Toward the structure of presenilin/gamma-secretase and presenilin homologs. Biochim Biophys Acta 1828(12):2886–2897
Acknowledgments
We are grateful to Dr. Andreas Klingl for assistance in electron microscopy and Prof. Uwe-G. Maier for allocation of the electron microscopic facilities. A.K. was supported by the LOEWE Research Centre for Synthetic Microbiology (Synmikro). A. L. H. was supported by the German Research Council within the framework of the Collaborative Research Center 987 “Microbial Diversity in Environmental Signal Response”. M. v. W. was supported by a grant from the German Research Council AL1206/3-1 and SVA received intramural funds from the Max Planck Society.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by M. W. W. Adams.
This article is part of a special issue based on the 10th International Congress on Extremophiles held in Saint Petersburg, Russia, September 7–11, 2014.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Henche, AL., van Wolferen, M., Ghosh, A. et al. Dissection of key determinants of cleavage activity in signal peptidase III (SPaseIII) PibD. Extremophiles 18, 905–913 (2014). https://doi.org/10.1007/s00792-014-0675-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-014-0675-4