
Abstract
Bacterial and archaeal community structures and diversity of three different sedimentary environments (BH1A, BH2A and BH3A) in the acid pit lake of a chalcopyrite mine at Touro (Spain) were determined by 16S rRNA gene PCR-DGGE and sequencing of clone libraries. DGGE of bacterial and archaeal amplicons showed that the sediments harbor different communities. Bacterial 16S rRNA gene sequences were assigned to Acidobacteria, Actinobacteria, Cyanobacteria, Planctomycetes, Proteobacteria, Chloroflexi and uncultured bacteria, after clustering into 42 operational taxonomic units (OTUs). OTU 2 represented approximately 37, 42 and 37 % of all sequences from sediments BH1A, BH2A and BH3A, respectively, and was phylogenetically related to uncultured Chloroflexi. Remaining OTUs were phylogenetically related to heterotrophic bacteria, including representatives of Ferrithrix and Acidobacterium genera. Archaeal 16S rRNA gene sequences were clustered into 54 OTUs. Most of the sequences from the BH1A sediment were assigned to Euryarchaeota, whereas those from BH2A sediment were assigned to Crenarchaeota. The majority of the sequences from BH3A sediment were assigned to unclassified Archaea, and showed similarities to uncultured and unclassified environmental clones. No sequences related to Acidithiobacillus and Leptospirillum, commonly associated with acid mine drainage, were detected in this study.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pit lakes are common in coal and metal sulfide open cut mining areas, and frequently originated from acid mine drainage (AMD), which is characteristically enriched in heavy metals such as Fe and Cu, and potentially toxic to the environment. AMD occurs when metal sulfide minerals (mostly pyrite, FeS2) are oxidized upon exposure to air and water, generating a solution with high concentrations of H+ and soluble metals (Nordstrom 1982). The rates of AMD generation may be affected by several factors, including the availability of oxidant and the microbial populations colonizing the mineral surfaces.
Overall, Eqs. (1)–(3) describe the oxidation of pyrite (Nordstrom 1982; Nordstrom and Alpers 1999):
When O2 is limiting and pH is low, ferric ion may oxidize pyrite generating more acidity (Eq. 4). In these environments, prokaryotic microorganisms, mostly chemoautotrophic, play important roles in the oxidation of reduced forms of sulfur, contributing to AMD generation.
AMD pit lakes are therefore extreme environments and may harbor unique microbial populations involved in the geochemistry of iron and sulfur. Several studies have been performed in the Iberian Pyritic Belt rivers, such as Tinto and Odiel, and in different AMDs in Spain and other parts of the world (Álvarez et al. 1993; Galán et al. 2003; Lee 2006; Nieto et al. 2007; Romero et al. 2007; Sánchez-Andrea et al. 2011; Sánchez-España et al. 2005) to understand metal geochemistry. However, studies on the microbial communities involved in the geochemistry of different metals in AMD systems, e.g., tailings, streams, biofilms, and pit lakes, are sparse (Auld et al. 2013; Hallberg et al. 2006; Rowe et al. 2007) and mostly concentrated in Chinese mines (Chen et al. 2013; Hao et al. 2010; Kuang et al. 2013; Yin et al. 2008).
The AMD pit lake of the Touro mine has been thoroughly chemically characterized. However, the microbial communities associated to the AMD pit lake sediments are unknown. Understanding the diversity of microorganisms that colonize AMD pit lakes and their functions may contribute to the development of new approaches for the bioremediation of AMD and AMD-contaminated sites. The goal of this study was to determine the bacterial and archaeal community structures and diversity in sediments from the AMD pit lake of the Touro mine and their possible roles in the geochemistry of iron and sulfur.
Materials and methods
Study area and sample collection
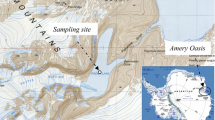
The study was carried out at the copper mine of Touro (Galicia, Spain, 42°52′24″N, 8°18′17″W) (Online Resource 1). The local geological substrate is formed mainly by schists and garnetiferous amphibolites, characterized by high concentrations of metal sulfides such as pyrite, pyrrhotite and chalcopyrite. The deposits of the metal sulfide are due to several volcanic intrusions into amphibolite. The spoil dump, mine slope and other surfaces of the quarry conserve a variable content of metal sulfide, even after Cu extraction (Álvarez et al. 2010; 2011; Otero et al. 2012). These deposits were exploited as an open cast mine, which occupies an area of 390 ha. The mine operated until the 1980s and at present the mine slopes and dumps are in process of restoration (Online Resource 1).
During the period of Cu extraction, the slurry materials were concentrated in a waste mud pile comprising a surface area of 80 ha and 80 m depth (Online Resource 1). The AMD (pH <3) was formed by the percolation of water through the pyrite-rich slurry materials and accumulated in a pit lake at the base of the mine. In the pit lake, several minerals precipitate forming sediments rich in Fe and trace metals (Online Resource 1).
Three types of sediments were sampled for microbial community analysis, according to their visual characteristics (Online Resource 1). The strong red precipitated sediment, named BH1A, and the red sediment with green filamentous organisms, named BH2A, were collected at the bottom of the lake, whereas a crusty superficial sediment, named BH3A, was sampled at the edge of the pit lake. The distance between each point was approximately 1 m and the depth of the water column oscillated between 0.5 and 1.0 m, except for the BH3A site, where the water column was approximately 0.10 m. The sediment samples were collected in sterile tubes in duplicates, freeze-dried and stored at −80 °C until processing.
Climate data was obtained from the meteorological station of Santiago de Compostela, located 10 km SW of the Touro mine (MeteoGalicia 2013). The average air temperature in the sampling day was 10.7 °C (day maximum 17.7 °C, minimum 6.0 °C). The average relative humidity was 62 % with no precipitation in the previous 15 days before sampling.
Sediment characterization
Sediment pH and redox potential (Eh) were measured in situ using a pH-redox meter (Hanna Instruments, model HI 9025). Final readings of redox potential were corrected by the addition of +244 mV of a calomel reference electrode. Sediment samples were centrifuged at 12,500g and 4 °C to extract porewater, and freeze-dried subsequently. In the porewater, the concentrations of dissolved Fe2+ and Fe3+ were determined using the 1,10-phenanthroline method (Stucki 1981). The following determinations were performed in the sediment solid fraction: pH in water (1:2.5, v:v), total organic carbon (TOC), total S (TS) and total nitrogen (TN) using a LECO CNS-2000 elemental analyzer. Total concentrations of Fe and Cu were determined after extraction of 0.5 g of ground sample with 15 ml of a mixture of HNO3:HCl:HF (9:3:3, v/v/v) and heating the mixture in a Ethos Plus (Milestone) microwave oven for 20 min (Otero et al. 2013). Exchangeable Fe and Cu were extracted with 1 M MgCl2 at pH 7 for 30 min (Tessier et al. 1979). Total and exchangeable metals were measured by graphite furnace atomic absorption spectrometry (Perkin-Elmer 4110ZL).
DNA extraction
Metagenomic DNA was extracted from 0.5 g of sediment sample. Two independent extractions per replicate were performed using the Fast DNA Spin kit (MP Biomedicals, France) and Fast Prep FP101 instrument (Savant, USA), according to the manufacturer’s instructions. DNA integrity was evaluated by electrophoresis in 1 % agarose gel run in 1× TAE buffer (Tris, Acetic Acid, EDTA) and stained with Sybr Green (Life Technologies, USA). Gel image capture was performed using a Storm densitometer (GE Healthcare, Brazil). DNA concentration was determined by fluorometry using a Qubit fluorometer (Life Technologies, USA) and the Quant-iT dsDNA BR kit (Life Technologies, USA).
Bacterial and archaeal 16S rRNA gene amplicon analyses
Equal amounts of metagenomic DNA extracted from the sediment samples (two replicates) were pooled and used as template for amplification. A fragment of the 16S rRNA gene was amplified from metagenomic DNA by polymerase chain reaction (PCR) using the universal primers PRBA338fGC (5′-CGC CCG CCG CGC GCG GCG GGC GGG GCG GGG GCA CGG GGG GAC TCC TAC GGG AGG C-3′) and PRUN518R (5′-ATT ACC GCG GCT GCT GG-3′) for Bacteria. For Archaea, the amplification was performed by nested PCR using primers ARCH 21F (5′-TTC YGG TTG ATC CYG CCR GA-3′, Y=C or T, R=A or G) and ARCH 958R (5′-YCC GGC GTT GAN TCC AAT T-3′) for the first amplification, and ARCH 340FGC (5′-CGC CCG CCG CGC GCG GCG GGC GGG GCG GGG GCA CGG GGG GCC CTA CGG GGY GCA SCA G-3′, Y=C or T, S=C or G) and ARCH 519R (5′-TTA CCG CGG CKG CTG-3′, K=G or T) for the second amplification.
Amplification reactions were performed in triplicate in 1× Taq Polymerase Buffer (Fermentas Life Sciences, Canada) containing 100 ng of template DNA, 1.5 mM MgCl2, 0.2 mM dNTPs (Life Technologies, USA), 1.5 U recombinant Taq DNA Polymerase (Fermentas Life Sciences, Canada), and 25 ρmol of each primer. PCR amplification conditions were 5 min at 95 °C; 30 cycles 1 min at 92 °C, 1 min at 55 °C and 1 min at 72 °C; and final extension for 10 min at 72 °C for bacterial DNA, and 5 min at 95 °C; 30 cycles of 30 s at 95 °C, 30 s at 55 °C and 1 min at 72 °C; and final extension for 10 min at 72 °C for archaeal DNA. DNA integrity and concentration were determined as described above.
Equal amounts of amplicons from three amplification reactions per sample were pooled and 300 ng analyzed by DGGE using 8 % (w/v) acrylamide:bisacrylamide (37.5:1, m:m) gels containing a 15–55 % linear gradient of formamide and urea (100 % denaturing solution contained 40 % formamide and 7 M urea) according to Muyzer et al. (1993). Electrophoresis was performed at 200 V constant and 60 °C, using a DCode System (BioRad, USA), in 1× TAE buffer. Gels were stained with Vistra Green (GE Healthcare Life Sciences, Brazil) and analyzed by densitometry, using a Storm densitometer (GE Healthcare Life Sciences, Brazil) and the program Diversity Database (BioRad, USA).
DGGE banding patterns representing the bacterial and archaeal community structures in the sediments were analyzed as discrete data (presence or absence of bands with the same mobility in the gel, Rf) using Hierarchical Clustering Analysis based on simple matching similarity matrices calculated with the Ward’s algorithm and Euclidian distances (Systat, SPSS Inc).
Bacterial and archaeal 16S rRNA gene sequencing
Metagenomic DNA from sediments BH1A, BH2A and BH3A was amplified by PCR using the primers BAC 8F (5′-AGA GTT TGA TCC TGG CTC AG-3′) and BAC 1541R (5′-AAG GAG GTG ATC CAG CCG CA-3′) for Bacteria, and ARCH 21F/ARCH 958R and ARCH 21F/ARCH 519R for Archaea. PCR conditions were as described above, except that the amount of primers was 10 ρmol each.
Amplicons were analyzed by electrophoresis on 1 % agarose gels in 1× TAE buffer and stained with Sybr Green (Life Technologies, USA). Amplicons with the expected sizes were purified with the Invisorb CleanUp kit (Invitek, Germany), and ligated to pGEM-T Easy vector (Promega, USA) at 4 °C overnight, according to the manufacturer’s instructions. The ligation product was transformed into E. coli DH5α competent cells by heat shock and transformed cells plated on LB-agar, containing ampicillin (100 μg mL−1) and X-Gal (5-Brome-4-chloro-3-indolyl-β-d-galactoside). A total of 192 colonies containing recombinant plasmids were selected for each sediment library and cells grown in liquid LB medium containing 100 μg ampicillin mL−1 medium at 37 °C overnight. Plasmids were extracted by the alkaline lyses method (Sambrook et al. 2001) and the inserts sequenced using an ABI 3100 Automatic Sequencer (Life Technologies, USA), the DYEnamic ET Terminator Cycle Sequencing Kit (GE Healthcare, Brazil) and primers for T7 (5′-TAA TAC GAC TCA CTA TAG GGC-3′) and SP6 (5′-ATT TAG GTG ACA CTA TAG AAT ACT C-3′) regions, following the manufacturer’s instructions.
Sequence analyses, operational taxonomic units (OTUs) definition and richness and diversity indices estimations
Nucleotide sequences (reads) were trimmed for the removal of low-quality bases (quality parameter >20, i.e., less than one error in 100 nucleotides) and vector sequences using Phred program (Ewing and Green 1998). Chimeric sequences were detected using the Chimera-Check algorithm (Cole et al. 2009) and excluded from the dataset. Valid sequences were aligned using ClustalX2 software (Thompson et al. 1997), setting gap opening penalty to 10 and gap extension penalty to 0.1 for pairwise and multiple alignments, and a Jukes–Cantor distance matrix was calculated by DNAdist (Felsenstein 1989). Sequences were clustered into OTUs using DOTUR (Schloss and Handelsman 2005), considering d = 0.03 for species definition. OTU richness was estimated using ACE and Chao-1 nonparametric estimators, and Shannon and reciprocal of Simpson’s diversity indices were estimated using SPADE (Chao and Shen 2003).
Taxonomical affiliation of the sequences was performed using the Classifier tool of the Ribosomal Database Project II (http://rdp.cme.msu.edu/), with a confidence level of 80 %, naïve Bayesian algorithm and taxonomic hierarchy RDP 16S rRNA training set 9 (Wang et al. 2007). The sequences were also compared with the National Center for Biotechnology Information (NCBI) nucleotide database (NT/NR) using the MegaBlast (Altschul et al. 1990). Neighbor-joining phylogenetic trees with the most representative OTUs were constructed based on Kimura-2 algorithm and 1,000 bootstrap replicates using MEGA 5 (Tamura et al. 2011). The best sequence match of each OTU in Megablast (database NT/NR) was included in the phylogenetic tree as a reference (Altschul et al. 1990).
Multiple comparisons among the 16S rRNA gene clone libraries were performed using the S-Libshuff algorithm (Schloss et al. 2004).
The nucleotide sequences used in this study have been deposited in the Genbank database under the accession numbers KC746537 to KC747003.
Results
Sediments characterization
The three sediments sampled showed extremely acidic and oxic conditions, with pH ranging from 2.2 to 3.5, redox potential higher than 700 mV, and high concentrations of Fe2+ and Fe3+ in the porewater (Table 1). In the sediment BH1A, the concentrations of TOC, TS and TN were lower than in BH2A and BH3A. Total concentrations of Fe ranged from 39 to 47 %, and Cu from 100 to 120 mg kg−1 (Table 1). The total Cu concentration in the sediments was 2–3 times higher than in natural soils over amphibolite rocks (49 ± 31 mg kg−1, according to Macías and Calvo de Anta 2009). The concentration of exchangeable Fe and Cu in the sediments was much lower than total concentration, but potentially more toxic since the exchangeable fraction presents high mobility and bioavailability in soils and sediments.
Bacterial community structure and diversity
Based on the PCR-DGGE banding patterns, the sediments sampled showed distinct bacterial community structures and the bacterial community of sediments, BH2A and BH3A were more similar to each other as compared to sediment BH1A (Fig. 1a).

Hierarchical clustering based on the profiles of bacterial (a) and archaeal (b) 16S rRNA gene amplicons from pit lake sediments BH1A, BH2A and BH3A
A total of 246 valid bacterial 16S rRNA gene sequences, 87 for BH1A, 76 for BH2A and 83 for BH3A, were obtained and used for further analyses. The majority of the sequences were assigned to the phylum Chloroflexi. Representatives of the phyla Actinobacteria, Planctomycetes and Proteobacteria were detected in all samples, whereas Acidobacteria was detected only in BH1A, and Cyanobacteria only in BH2A and BH3A. Nearly 6 % of BH1A and BH3A sequences were classified only at the Bacteria domain level (Table 2).
Bacterial richness (Chao-1 and ACE estimators) and diversity (Shannon and reciprocal of Simpson’s) indices were not statistically different among the three sediments (P < 0.05) (Table 2), even though the structures of their communities were statistically different, based on S-Libshuff analyses (P = 0.01) (data not shown). Calculated sampling coverage was in the range of 88–90 %, although rarefaction curves suggested the need of an additional sequencing effort for covering most of the bacterial diversity (Online Resource 2a).
Valid sequences were clustered into 42 OTUs (d = 0.03). The complete list of the sequences comprising each OTU is shown in Online Resource 3. Only 6 OTUs were shared among all libraries and 9, 7 and 13 OTUs were exclusive of libraries BH1A, BH2A and BH3A, respectively. Phylogenetic analyses indicated the presence of a small dominant group of bacteria (Fig. 2) represented by OTU 2, which showed approximately 37, 42 and 37 % abundance in sediments BH1A, BH2A and BH3A, respectively. The clade containing OTUs 2, 3, 4, 5, 6, 10, 17 and 19 represented approximately 82, 56 and 64 % of the valid sequences for sediments BH1A, BH2A and BH3A, respectively (Fig. 2), and showed similarity to uncultured bacteria from the Nanshan waste ore deposits on the Mountain Xiang in China (access DQ453119, Hao et al. 2007), a volcanic deposit in Hawaii (AY917600, Gomez-Alvarez et al. 2007), unvegetated soil environments on Signy Island in Antartic (EF221273, Yergeau et al. 2007), and Ktedonobacteria Hsw-67 (Chloroflexi) from underground coal mine fire vent soil in China (GU237174, Xu and Zeng 2009, unpublished).

Phylogenetic tree of bacterial 16S rRNA gene sequences from pit lake sediments BH1A, BH2A and BH3A. Tree was constructed using neighbor-joining. Bootstrap values higher than 50 % based on 1,000 replicates are indicated at the nodes. Scale bar represents 5 % nucleotide substitutions per position. Best match sequences from NCBI database were used as references. Values in the parentheses correspond to the relative abundance (%) of the OTU in BH1A, BH2A and BH3A sediment sample, respectively
Less representative OTUs were mainly related to heterotrophic bacteria. OTUs 1 and 28, representing 14 % of BH2A sequences, showed similarity to Mycobacterium sp (AF236834, Leclerc et al. 2000) and Mycobacterium shimoidei (X82459, Boettger 2003, unpublished). OTU 22, representing 9 % of the BH2A sequences, was phylogenetically related to an uncultured Planctomycetacia (EF073417, Jangid et al. 2008). OTUs 29 and 36, representing 6 % of BH3A sequences, showed similarity to an uncultured Gemmata from sediments of an acid mine pit lake (FJ228382, Meier et al. 2009, unpublished). OTUs 27, 30 and 31, representing approximately 1 and 7 % of BH2A and BH3A sequences, respectively, showed similarity to an uncultured cyanobacterium clone (EF219700, Yergeau et al. 2007).
OTU 21 (5 % of the BH2A sequences) showed similarity to the obligate heterotrophic Ferrithrix thermotolerans (AY140237, Johnson et al. 2003), which is able to accelerate the oxidation of pyrite in the presence of yeast extract and dissimilatory reduction of ferric iron under anoxigenic conditions (Johnson et al. 2009) at moderate thermophilic conditions. This OTU also showed similarity to an uncultured bacterium from copper mine AMD (DQ458034). OTU 16 (2.3 % of the BH1A sequences) showed similarity to Acidobacterium capsulatum (CP001472, Ward et al. 2009), an acidophilic chemoorganotrophic isolated from acidic mineral environments (Kishimoto et al. 1991).
None of the detected sequences showed similarity to bacteria commonly associated with AMD, such as Acidithiobacillus and Leptospirillum.
Archaeal community structure and diversity
Similar to bacteria, the sediments showed distinct archaeal community structures, based on PCR-DGGE banding patterns (Fig. 1b). However, the levels of dissimilarity were higher, as compared to the ones observed for the bacterial communities. Similar results were observed by 16S rRNA gene cloning and sequencing. S-Libshuff analysis (P = 0.01) showed that the archaeal communities in the sampled sediments were statistically different (data not shown).
A total of 221 valid archaeal 16S rRNA gene sequences, 85 for BH1A, 73 for BH2A and 63 for BH3A, were obtained and used for further analyses. Approximately 58 % of the BH1A sequences were assigned to the phylum Euryarchaeota and 92 % of the BH2A sequences to Crenarchaeota. The remaining sequences, including approximately 98 % of BH3A sequences, showed no similarities to other sequences in the databases and were assigned to unclassified Archaea (Table 2).
Richness (Chao-1 and ACE estimators) and the Shannon diversity index were statistically lower in BH2A, as compared to BH1A and BH3A (Table 2). Estimated sample coverage ranged from approximately 76 % (BH3A) to 98 % (BH2A). Rarefaction curves indicated that most of the archaeal species were sampled (Online Resource 2b).
The valid sequences were clustered into 54 OTUs (d = 0.03). The complete list of sequences comprising each OTU is shown in Online Resource 3. All OTUs from BH2A sediment were clustered in a single clade and showed similarity to an uncultured Crenarchaeotum from a tidal flat sediment (AY396690, Kim et al. 2005), uncultured archaea from marine sediments (access DQ988474, GQ927558, GQ926246, Hu et al. 2006, unpublished) and a methane seep river sediment (FJ264795, Beal et al. 2009). Together, OTUs 29 and 30 represented approximately 77 % of all BH2A sequences (Fig. 3). The clade containing OTUs 3, 4, 6, 19, 20 and 21, represented approximately 33 % of BH1A sequences, and showed similarity to an uncultured archaeon from forest wetland impacted by rejected coal (AF523938, Brofft et al. 2002) and an uncultured Thermoplasmatales (Euryarchaeota) from sediments of an acid mine pit lake (FJ228375, Meier et al. 2009, unpublished). OTUs 7, 24 and 25, representing approximately 25 % of BH1A sequences, showed similarity to uncultured archaea from a methane seep (GQ356875, Beal et al. 2005, unpublished) and volcano sediment underneath an iron-oxidizing mat (EF687634, Omoregie et al. 2008). OTU 36, representing approximately 36 % of BH3A sequences, showed similarity to uncultured archaea from an acidic red soil (FJ174719, Ying et al. 2010) and a rhizospheric soil (EF020979, Lesaulnier et al. 2008).

Phylogenetic tree of archaeal 16S rRNA gene sequences from pit lake sediments BH1A, BH2A and BH3A. Tree was constructed using neighbor-joining. Bootstrap values higher than 50 % based on 1,000 replicates are indicated at the nodes. Scale bar represents 5 % nucleotide substitutions per position. Best match sequences from NCBI database were used as references. Values in the parentheses correspond to the relative abundance (%) of the OTU in BH1A, BH2A and BH3A sediment sample, respectively
Discussion
The mining process at the Touro mine consisted basically in separating out chalcopyrite from pyrite, which becomes one of the major components of the slurry. The abiotic and biotic oxidation of pyrite (FeS2) are the main geochemical processes that contribute to water acidification and increased solubility of Fe and Cu under oxic conditions (Álvarez et al. 1993). Pyrite can be oxidized by oxygen and ferric iron regenerated by microorganisms that catalyzes the oxidation of ferrous ions (Fowler et al. 1999; Silverman and Ehrlich 1964). The oxidation of Fe(II) to Fe(III) and reduced forms of sulfur by chemoautotrophic microorganisms results in proton generation and consequent acidification of mine drainage water, which has to be properly treated and disposed (Baker and Banfield 2003).
In our study, the exchangeable and total concentrations of Fe in the pit lake sediments indicated a strong influence of the percolation water from the slurry materials, which were enriched in pyrite and depleted in chalcopyrite due to the previous extraction of the former. Hence, high concentrations of dissolved Fe in the porewater were observed. In addition, due to oxic and extremely acidic conditions, the concentrations of Fe3+ were higher than Fe2+ in porewater. In contrast, the total concentration of Cu was lower, and the results of the sequential extraction showed that most of the Cu was associated with Fe-oxyhydroxides (data not shown) and, therefore, may not be readily bioavailable.
The physical–chemical analysis of the Touro AMD pit lake sediments also reveled higher concentrations of TOC, TS and TN in samples BH2A and BH3A than in BH1A. These results may be due to the presence of a layer of algae related to Zygnematales, based on the sequence of chloroplastic 16S rRNA gene (data not show), in BH2A or arrival of organic matter from the edges of the pit lake in BH3A (Online Resource 1).
In AMD, the richness of microbial species is normally low and limited by the number of possible energy-deriving reactions (Baker and Banfield 2003). In our study, the bacterial 16S rRNA gene sequences obtained were clustered into 42 OTUs assigned to Acidobacteria, Actinobacteria, Cyanobacteria, Planctomycetes, Proteobacteria, Chloroflexi and unclassified bacteria. The bacterial richness observed was comparable to the one observed in wetland impacted with rejected coal pile drainage water (Brofft et al. 2002), and abandoned lead–zinc mine tailings (Zhang et al. 2007), but greater than the richness observed in sulfide AMD (He et al. 2007) and in macroscopic filaments from the Tinto River (Spain) (García-Moyano et al. 2007).
The microbial community associated with the sediments of the Touro mine AMD pit lake, in our study, was highly dominated by few bacterial groups, since 19 % of the OTUs corresponded to approximately 82, 56 and 64 % of all 16S rRNA gene clone sequences from BH1A, BH2A and BH3A, respectively. The dominance of few bacterial species may be attributed to the acid and energetically limited mineral substrates in such an environment where autotrophic metabolism is predominant. The phylogenetic analysis of the dominant OTUs (Fig. 2) indicated similarities to Chloroflexi-related uncultured bacteria from AMD impacted environments and volcanic areas. The phylum Chloroflexi is ubiquitous and a limited number of cultured representatives is known (Yamada and Sekiguchi 2009). High dominance of Choroflexi-like organisms has also been observed in a Fe(III)-rich sediment from an AMD impacted area in the USA (Senko et al. 2008). However, the roles of Chloroflexi in such environments are still uncertain. Species such as Chloroflexus spp. and Roseiflexus spp. may inhabit microbial mats in neutral and alkaline geothermal springs and alternate between heterotrophic and photosynthetic metabolism (Klatt et al. 2013; Zarzycki et al. 2009). In addition, carbon fixation by acidophilic filamentous algae, such as Zygnematales, would contribute to the growth of heterotrophic iron reducers, as proposed by Rowe et al. (2007). Recently, Nancucheo and Barrie Johnson (2012) observed that heterotrophic acidophilic Acidiphilium and Acidobacterium spp. are able to metabolize monosaccharides produced by Chlorella and Euglena in AMD environments. Souza-Egipsy et al. (2008) proposed that photosynthetic biofilms in the Tinto River might be involved either in iron oxidation, through the release of oxygen in the water, or reduction, providing organic carbon for iron-reducing heterotrophic acidophiles. The role of algae biofilms on iron metabolism in the Touro AMD pit lake sediments, however, is not known and further studies on the eukaryotic diversity and function would be necessary.
The occurrence of the obligate chemolithotrophic bacteria, such as Acidithiobacillus ferrooxidans, A. thiooxidans and Leptospirillum ferrooxidans, has been frequently reported in AMD (Schippers and Sand 1999; Schrenk et al. 1998). However, in our study, no sequences phylogenetically related to these genera have been detected, suggesting that they do not occur in these sediments or that their abundances are below the detection limits of the methodological approach used. It is possible that in AMDs with pH >3, moderately acidophilic iron/sulfur oxidizing bacteria play a major role in metal oxidation, minimizing the importance of extremely acidophilic oxidizers (Hallberg and Johnson 2003). The absence of A. ferrooxidans and L. ferrooxidans might also be attributed to the depletion of their substrates such as Fe(II) (Table 2). Corroborating this hypothesis, the sequential extraction of iron in our study showed that Fe-oxyhydroxides are more abundant than Fe-pyrite. Furthermore, in such environments, pyrite might be coated with the Fe-oxyhydroxides precipitates, limiting the access of bacteria to pyrite surface and the oxidation process (Huminicki and Rimstidt 2009).
In contrast to Bacteria, most Archaea detected in our study showed no similarity to known species, including genera frequently reported in AMD such as Sulfolobus, Acidianus and Ferroplasma (Baker and Banfield 2003). However, the archaeal 16S rRNA gene sequences detected in our study were similar to sequences detected in soil, deep sea, marine sediments, volcanic areas and AMD impacted sites, indicating the dominance of so far uncultured archaeal species commonly detected in environments under extreme conditions. From the available data, the functions of the archaeal species in the sediments of the Touro pit lake cannot be established, and further studies would be necessary to determine their functions in this environment.
Conclusion
The study of the prokaryotic diversity in the AMD pit lake of Touro is essential for the understanding of the geochemical transformations taking place and developing of new approaches for AMD remediation. Regardless of the spatial proximity, the sediments studied showed distinct bacterial and archaeal communities. A high dominance of Chloroflexi-like microorganisms was observed. Even though they may act as primary producers, contributing to the heterotrophic growth of acidophilic microorganisms, their functions still remain unclear. Most archaeal sequences showed similarity to uncultured and unidentified environmental archaea and their possible functions cannot be established.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Álvarez E, Pérez A, Calvo R (1993) Aluminum speciation in surface waters and soils solutions in areas of sulphide mineralization in Galicia (NW Spain). Sci Total Environ 133:17–37
Álvarez E, Fernandez-Sanjurjo MJ, Otero XL, Macias F (2010) Aluminum geochemistry in the bulk and rhizospheric soil in the species colonising and abandoned copper mine in Galicia (NW Spain). J Soil Sediment 10:1236–1245
Álvarez E, Fernandez-Sanjurjo MJ, Otero XL, Macias F (2011) Aluminum speciation in the bulk and rhizospheric soil solution of the species colonizing and abandoned copper mine in Galicia (NW Spain). J Soil Sediment 11:221–230
Auld RR, Myre M, Mykytczuk NC, Leduc LG, Merritt TJ (2013) Characterization of the microbial acid mine drainage microbial community using culturing and direct sequencing techniques. J Microbiol Methods 93:108–115
Baker BJ, Banfield JF (2003) Microbial communities in acid mine drainage. FEMS Microbiol Ecol 44:139–152
Beal EJ, House CH, Orphan VJ (2009) Manganese- and iron-dependent marine methane oxidation. Science 325:184–187
Brofft JE, McArthur JV, Shimkets LJ (2002) Recovery of novel bacterial diversity from a forested wetland impacted by reject coal. Environ Microbiol 4:764–769
Chao A, Shen TJ (2003) Nonparametric estimation of Shannon’s index of diversity when there are unseen species in sample. Environ Ecol Stat 10:429–443
Chen LX, Li JT, Chen YT, Huang LN, Hua ZS, Hu M, Shu WS (2013) Shifts in microbial community composition and function in the acidification of a lead/zinc mine tailings. Environ Microbiol. doi:10.1111/1462-2920.12114
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje MJ (2009) The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37:D141–D145
Ewing B, Green P (1998) Base-calling of automated sequencer traces using Phred. II error probabilities. Genome Res 8:186–194
Felsenstein J (1989) PHYLIP—phylogeny inference package (Version 3.2). Cladistics 5:164–166
Fowler TA, Holmes PR, Crundwell FK (1999) The mechanism of bacterial leaching of pyrite by Thiobacillus ferrooxidans. Appl Environ Microbiol 65:2987–2993
Galán E, Gómez-Ariza JL, González I, Fernández-Caliani JC, Morales E, Giráldez I (2003) Heavy metals partitioning in river sediments severely polluted by acid mine drainage in the Iberian Pyrite Belt. App Geochem 18:409–421
García-Moyano A, González-Toril E, Aguilera A, Amils R (2007) Prokaryotic community composition and ecology of floating macroscopic filaments from an extreme acid environment, Río Tinto (SW, Spain). Syst Appl Microbiol 30:601–614
Gomez-Alvarez V, King GM, Nüsslein K (2007) Comparative bacterial diversity in recent Hawaiian volcanic deposits of different ages. FEMS Microbiol Ecol 60:60–73
Hallberg KB, Johnson DB (2003) Novel acidophiles isolated from moderately acidic mine drainage waters. Hydrometallurgy 71:139–148
Hallberg KB, Coupland K, Kimura S, Johnson DB (2006) Macroscopic streamer growths in acidic, metal-rich mine waters in north Wales consist of novel and remarkably simple bacterial communities. Appl Environ Microbiol 72:2022–2030
Hao CB, Zhang HX, Bai ZH, Hu Q, Zhang BG (2007) A novel acidophile community populating waste ore deposits at an acid mine drainage site. J Environ Sci 19:444–450
Hao C, Wang L, Gao Y, Zhang L, Dong H (2010) Microbial diversity in acid mine drainage of Xiang Mountain sulfide mine, Anhui Province, China. Extremophiles 14:465–474
He Z, Xiao S, Xie X, Zhong H, Hu Y, Li Q, Gao F, Li G, Liu J, Qiu G (2007) Molecular diversity of microbial community in acid mine drainages of Yunfu sulfide mine. Extremophiles 11:305–314
Huminicki DMC, Rimstidt JD (2009) Iron oxyhydroxide coating of pyrite for acid mine drainage control. App Geochem 24:1626–1634
Jangid K, Williams MA, Franzluebbers AJ, Sanderlin JS, Reeves JH, Jenkins MB, Endale DM, Coleman DC, Whitman WB (2008) Relative impacts of land-use, management intensity and fertilization upon soil microbial community structure in agricultural systems. Soil Biol Biochem 40:2843–2853
Johnson DB, Okibe N, Roberto FF (2003) Novel thermo-acidophilic bacteria isolated from geothermal sites in Yellowstone National Park: physiological and phylogenetic characteristics. Arch Microbiol 180:60–68
Johnson DB, Bacelar-Nicolau P, Okibe N, Thomas A, Hallberg KB (2009) Ferrimicrobium acidiphilum gen. nov., sp. nov. and Ferrithrix thermotolerans gen. nov., sp. nov.: heterotrophic, iron-oxidizing, extremely acidophilic actinobacteria. Int J Syst Evol Microbiol 59:1082–1089
Kim BS, Oh HM, Kang H, Chun J (2005) Archaeal diversity in tidal flat sediment as revealed by 16S rDNA analysis. J Microbiol 43:144–151
Kishimoto N, Kosako Y, Tano T (1991) Acidobacterium capsulatum gen. nov., sp. nov.: an acidophilic chemoorganotrophic bacterium containing menaquinone from acidic mineral environment. Current Microbiol 22:1–7
Klatt CG, Liu Z, Ludwig M, Kühl M, Jensen SI, Bryant DA, Ward DM (2013) Temporal metatranscriptomic patterning in phototrophic Chloroflexi inhabiting a microbial mat in a geothermal spring. ISME J. doi:10.1038
Kuang JL, Huang LN, Chen LX, Hua ZS, Li SJ, Hu M, Li JT, Shu WS (2013) Contemporary environmental variation determines microbial diversity patterns in acid mine drainage. ISME J 7:1038–1050
Leclerc MC, Haddad N, Moreau R, Thorel MF (2000) Molecular characterization of environmental mycobacterium strains by PCR-restriction fragment length polymorphism of hsp65 and by sequencing of hsp65, and of 16S and ITS1 rDNA. Res Microbiol 151:629–638
Lee S (2006) Geochemistry and partitioning of trace metals in paddy soils affected by metal mine tailings in Korea. Geoderma 135:26–37
Lesaulnier C, Papamichail D, McCorkle S, Ollivier B, Skiena S, Taghavi S, Zak D, van der Lelie D (2008) Elevated atmospheric CO2 affects soil microbial diversity associated with trembling aspen. Environ Microbiol 10:926–941
Macías F, Calvo de Anta R (2009) Niveles genéricos de referencia de metales pesados y otros elementos traza en suelos de Galicia. Xunta de Galicia. http://solos.medioambiente.xunta.es/solos/documents/librongr.pdf. Accessed 23 March 2013
MeteoGalicia (2013) Consellería de Medio Ambiente, Territorio e Infraestruturas—Xunta de Galicia. http://www2.meteogalicia.es/galego/observacion/estacions/estacionsHistorico.asp?Nest=10124&prov=A%20Coru%F1a&red=102. Accessed 29 May 2013
Muyzer G, Dewaal EC, Uitterlinden AG (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16s rRNA. Appl Environ Microbiol 59:695–700
Nancucheo I, Barrie Johnson D (2012) Acidophilic algae isolated from mine-impacted environments and their roles in sustaining heterotrophic acidophiles. Front Microbiol 3:1–8
Nieto JM, Sarmiento AM, Olías M, Canovas CR, Riba I, Kalman J, Delvalls TA (2007) Acid mine drainage pollution in the Tinto and Odiel Rivers (Iberian Pyrite Belt, SW Spain) and bioavailability of the transported metals to the Huelva estuary. Environ Int 33:445–455
Nordstrom DK (1982) Aqueous pyrite oxidation and the consequent formation of secondary iron minerals. In: Kittrick JS, Fanning, DS, Hosser, LR (eds) Acid sulphate weathering. SSSA Special Publication Lumber 10, Soil Science Society of America, Madison, pp 37–55
Nordstrom DK, Alpers CN (1999) Geochemistry of acid mine waters. In: Plumlee GS, Logsdon, MJ (eds) The environmental geochemistry of mineral deposits. Part A: processes, techniques, and health issues. The Society of Economic Geologists, Littleton, pp 133–160
Omoregie EO, Mastalerz V, de Lange G, Straub KL, Kappler A, Røy H, Stadnitskaia A, Foucher JP, Boetius A (2008) Biogeochemistry and community composition of iron- and sulfur-precipitating microbial mats at the Chefren mud volcano (Nile Deep Sea Fan, Eastern Mediterranean). Appl Environ Microbiol 74:3198–3215
Otero XL, Alvarez E, Fernandez-Sanjurjo MJ, Macias F (2012) Micronutrients and toxic trace metals in the bulk and rhizospheric soil of the spontaneous vegetation at an abandoned copper mine in Galicia (NW Spain). J Geochem Explor 112:84–92
Otero XL, Huerta-Díaz MA, De La Peñas S, Ferreira TO (2013) Sand as a relevant fraction in geochemical studies in intertidal environments. Environ Monit Assess. doi:10.1007/s10661-013-3146-y
Romero FM, Armienta MA, Gónzalez-Hernández G (2007) Solid-phase control on the mobility of potentially toxic elements in an abandoned lead/zinc mine tailings impoundment, Taxco, Mexico. App Geochem 18:109–127
Rowe OF, Sánchez-España J, Hallberg KB, Johnson DB (2007) Microbial communities and geochemical dynamics in an extremely acidic, metal-rich stream at an abandoned sulfide mine (Huelva, Spain) underpinned by two functional primary production systems. Environ Microbiol 9:1761–1771
Sambrook J, Fritsch EF, Maniatis T (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, New York
Sánchez-Andrea I, Rodríguez N, Amils R, Sanz JL (2011) Microbial diversity in anaerobic sediments at Rio Tinto, a naturally acidic environment with a high heavy metal content. Appl Environ Microbiol 77:6085–6093
Sánchez-España J, López E, Santofimia E, Aduvire O, Reyes J, Barettino D (2005) Acid mine drainage in the Iberian Pyrite Belt (Odiel river watershed, Huelva, SW Spain): geochemistry, mineralogy and environmental implications. Appl Geochem 20:1320–1356
Schippers A, Sand W (1999) Bacterial leaching of metal sulfides proceeds by two indirect mechanisms via thiosulfate or via polysulfides and sulfur. Appl Environ Microbiol 65:319–321
Schloss PD, Handelsman J (2005) Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71:1501–1506
Schloss PD, Larget BR, Handelsman J (2004) Integration of microbial ecology and statistics: a test to compare gene libraries. Appl Environ Microbiol 70:5485–5492
Schrenk MO, Edwards KJ, Goodman RM, Hamers RJ, Banfield JF (1998) Distribution of Thiobacillus ferrooxidans and Leptospirillum ferrooxidans: implications for generation of acid mine drainage. Science 279:1519–1522
Senko JM, Wanjugi P, Lucas M, Bruns MA, Burgos WD (2008) Characterization of Fe(II) oxidizing bacterial activities and communities at two acidic Appalachian coalmine drainage-impacted sites. ISME J 2:1134–1145
Silverman MP, Ehrlich HL (1964) Microbial formation and degradation of minerals. In: Umbreit WW (ed) Advances in applied microbiology. Academic Press, London, pp 153–206
Souza-Egipsy V, González-Toril E, Zettler E, Amaral-Zettler L, Aguilera A, Amils R (2008) Prokaryotic community structure in algal photosynthetic biofilms from extreme acidic streams in Río Tinto (Huelva, Spain). Int Microbiol 11:251–260
Stucki JW (1981) The quantitative assay of minerals for Fe2+ and Fe3+ using 1,10-phenantroline. II. A photochemical method. Soil Sci Soc Am J 45:638–641
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Tessier A, Campbell PGC, Bisson M (1979) Sequential extraction procedure for the speciation of particulate trace metals. Anal Chem 5:844–855
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Ward NL, Challacombe JF, Janssen PH, Henrissat B, Coutinho PM, Wu M, Xie G, Haft DH, Sait M, Badger J, Barabote RD, Bradley B, Brettin TS, Brinkac LM, Bruce D, Creasy T, Daugherty SC, Davidsen TM, DeBoy RT, Detter JC, Dodson RJ, Durkin AS, Ganapathy A, Gwinn-Giglio M, Han CS, Khouri H, Kiss H, Kothari SP, Madupu R, Nelson KE, Nelson WC, Paulsen I, Penn K, Ren Q, Rosovitz MJ, Selengut JD, Shrivastava S, Sullivan SA, Tapia R, Thompson LS, Watkins KL, Yang Q, Yu C, Zafar N, Zhou L, Kuske CR (2009) Three genomes from the phylum Acidobacteria provide insight into the lifestyles of these microorganisms in soils. Appl Environ Microbiol 75:2046–2056
Yamada T, Sekiguchi Y (2009) Cultivation of uncultured Chloroflexi Subphyla: significance and ecophysiology of formerly uncultured Chloroflexi ‘Subphylum I’ with natural and biotechnological relevance. Microbes Environ 24:205–216
Yergeau E, Newsham KK, Pearce DA, Kowalchuk GA (2007) Patterns of bacterial diversity across a range of Antarctic terrestrial habitats. Environ Microbiol 9:2670–2682
Yin H, Cao L, Xie M, Chen Q, Qiu G, Zhou J, Wu L, Wang D, Liu X (2008) Bacterial diversity based on 16S rRNA and gyrB genes at Yinshan mine, China. Syst Appl Microbiol 31:302–311
Ying JY, Zhang LM, He JZ (2010) Putative ammonia-oxidizing bacteria and archaea in an acidic red soil with different land utilization patterns. Environ Microbiol Rep 2:304–312
Zarzycki J, Brecht V, Müller M, Fuchsa G (2009) Identifying the missing steps of the autotrophic 3-hydroxypropionate CO2 fixation cycle in Chloroflexus aurantiacus. Proc Natl Acad Sci USA 106:21317–21322
Zhang HB, Shi W, Yang MX, Sha T, Zhao ZW (2007) Bacterial diversity at different depths in lead-zinc mine tailings as revealed by 16S rRNA gene libraries. J Microbiol 45:479–484
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. da Costa.
Electronic supplementary material
Below is the link to the electronic supplementary material.
792_2013_576_MOESM1_ESM.jpg
{kind=link}
Online Resource 1 Details of the pit lake of the Touro mine collecting the AMD and sediment characteristics. Samples: BH1A. BH2A. BH3A (jpg 995 kb)
792_2013_576_MOESM2_ESM.tif
Online Resource 2 Rarefaction curves representing the estimated number of bacterial (a) and archaeal (b) OTUs as a function of the sequence effort, considering a cut-off evolutionary distance of 0.03. Bars represent 95 % confidence intervals (TIFF 17820 kb)
792_2013_576_MOESM3_ESM.pdf
Online Resource 3 List of sequences comprising each bacterial and archaeal OTU from sediments BH1A, BH2A and BH3A. A cut-off evolutionary distance of 0.03 was used to define OTU (PDF 407 kb)
Rights and permissions
About this article
Cite this article
Lucheta, A.R., Otero, X.L., Macías, F. et al. Bacterial and archaeal communities in the acid pit lake sediments of a chalcopyrite mine. Extremophiles 17, 941–951 (2013). https://doi.org/10.1007/s00792-013-0576-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-013-0576-y