
Abstract
The endogenous components of the thioredoxin system in the Antarctic eubacterium Pseudoalteromonas haloplanktis have been purified and characterised. The temperature dependence of the activities sustained by thioredoxin (PhTrx) and thioredoxin reductase (PhTrxR) pointed to their adaptation in the cold growth environment. PhTrxR was purified as a flavoenzyme and its activity was significantly enhanced in the presence of molar concentration of monovalent cations. The energetics of the partial reactions leading to the whole electron transfer from NADPH to the target protein substrate in the reconstituted thioredoxin system was also investigated. While the initial electron transfer from NADPH to PhTrxR was energetically favoured, the final passage to the heterologous protein substrate enhanced the energetic barrier of the whole process. The energy of activation of the heat inactivation process essentially reflected the psychrophilic origin of PhTrxR. Vice versa, PhTrx possessed an exceptional heat resistance (half-life, 4.4 h at 95 °C), ranking this protein among the most thermostable enzymes reported so far in psychrophiles. PhTrxR was covalently modified by glutathione, mainly by its oxidised or nitrosylated forms. A mutagenic analysis realised on three non catalytic cysteines of the flavoenzyme allowed the identification of C303 as the target for the S-glutathionylation reaction.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Proteins outside the cell or located on its surface are rich in disulphides, reflecting the oxidising conditions of the extracellular environment. In contrast, intracellular proteins containing disulphide bridges are rare because of the reducing properties of the cytosol (Gilbert 1990). The redox state of cysteine residues in intracellular proteins is controlled through thiol-disulphide exchange reactions triggered by specific enzyme systems displaying a fast and readily reversible activity (Holmgren 1985). This thiol redox control becomes crucial when the target cysteines have a specific role; indeed, this system is emerging as a major regulatory mechanism in either signal transduction or preservation of the reduced state of cytosolic proteins (Gilbert 1990). The thioredoxin system, composed by thioredoxin (Trx), thioredoxin reductase (TrxR) and NADPH is a key thiol-disulphide exchanger involved in maintaining target cytoplasmic proteins in their reduced state (Arnér and Holmgren 2000; Holmgren 1989). Both Trx and TrxR possess a conserved sequence with two neighbouring cysteines forming a reversible disulphide bridge essential for their functioning as redox exchangers. Indeed, TrxR catalyses the NADPH-dependent electrons transfer to the active disulphide of the oxidised form of Trx (Trx-S2) to form a dithiol (reaction 1 in the mechanism shown below); then, the reduced form of Trx (Trx-(SH)2) reduces the disulphide bridges eventually formed in target proteins (reaction 2).
Trx, conserved among all organisms, is a small protein, with a typical CXXC active-site motif. The first target proteins of the Trx activity, identified in bacterial and eukaryotic cells, are ribonucleotide reductase, 3′-phosphoadenosine-5′-phosphosulphate reductase and methionine-sulphoxide reductase (Tsang and Schiff 1976; Ejiri et al. 1979). Furthermore, it has been shown that Trx is involved in the activation of DNA binding activity of transcription factors (Matthews et al. 1992) and that a dimeric form of Trx binds to and improves the high processivity of T7 DNA polymerase (Huber et al. 1987).
TrxR is a homodimeric enzyme containing an active-site motif similar to that of Trx. The enzyme binds one molecule of FAD in each subunit and uses NADPH as electron donor (Moore et al. 1964). The activity of TrxR is essential for ribonucleotide metabolism (Holmgren 1989), regulation of transcriptional activity (Schenk et al. 1994) and protein folding (Kern et al. 2003). On the basis of the differences in size, structure and catalytic mechanism, TrxR is usually grouped in two classes (Arscott et al. 1997): type I high molecular mass TrxR (55–58 kDa per subunit), isolated from higher eukaryotes (Gasdaska et al. 1999; Kanzok et al. 2001), and type II low molecular mass TrxR (around 35 kDa per subunit), isolated from lower eukaryotes (Chae et al. 1994) and prokaryotes (Williams 1995). Both types of TrxR are members of the large family of pyridine nucleotide disulphide oxidoreductases, including lipoamide dehydrogenase, glutathione reductase and mercuric reductase (Ghisla and Massey 1989).
In recent years, the mechanisms leading to the formation/breakdown of disulphide bridges in proteins have been examined also in extremophilic organisms (Jeon and Ishikawa 2002; Kashima and Ishikawa 2003; Ruocco et al. 2004; Ladenstein and Ren 2006; Grimaldi et al. 2008; Hernandez et al. 2008; Cotugno et al. 2009). In particular, the components of the thioredoxin system have been characterised in some (hyper)thermophilic archaea, such as Sulfolobus solfataricus (Ruocco et al. 2004; Grimaldi et al. 2008), Aeropyrum pernix (Jeon and Ishikawa 2002), Pyrococcus horikoshii (Kashima and Ishikawa 2003) and Thermoplasma acidophilum (Hernandez et al. 2008). The information available for the thioredoxin system in cold-adapted microorganisms is limited to a recent paper, describing the properties of recombinant forms of Trx and TrxR from the psychrotolerant eubacterium Pseudoalteromonas haloplanktis TAC 125 (rPhTrx and rPhTrxR, respectively; Cotugno et al. 2009). It is known that this microorganism, isolated from the Antarctic sea and able to grow in a broad range of temperatures (4–20 °C), adapts its biochemical machinery to functioning either in the cold or in moderate temperature conditions (Birolo et al. 2000; Medigue et al. 2005). In particular, P. haloplanktis is adapted to fast growth, because the marine habitat, which is full of plankton debris, represents a rich medium; furthermore, in laboratory conditions, it grows to very high density in the presence of sufficient nutrients and high aeration, thus indicating that respiration is particularly efficient in this bacterium and that protection against reactive oxygen species must be ensured (Medigue et al. 2005). P. haloplanktis is also well adapted to salts because of its marine habitat, and some enzymes isolated from this bacterium show a marked halophilicity (Srimathi et al. 2007; Evangelista et al. 2009). All these findings make P. haloplanktis a valuable model to study the environment-driven adaptation mechanisms. Concerning the thioredoxin system in P. haloplanktis, the analysis of the effect of temperature on the structure–function relationships of rPhTrx and rPhTrxR indicated a differential cold adaptation among these enzymes (Cotugno et al. 2009). A structural reason for cold adaptation of psychrophilic proteins was suggested on the basis of their amino acid composition, as psychrophilic proteins use smaller-size and less hydrophobic amino acid residues, compared to the corresponding mesophilic counterparts (De Vendittis et al. 2008).
To further investigate the cold adaptation of the thioredoxin system, this work describes the properties of the components of this system isolated from P. haloplanktis cell extracts. The different psychrophilic character of the purified proteins (PhTrx and PhTrxR) has been assessed. In addition, the study was focused on the effect of different monovalent cations on the activity of PhTrxR. Furthermore, it was found that this enzyme is the target of a S-glutathionylation reaction, which, however, does not greatly affect its activity. Finally, a mutagenic analysis allowed the identification of the cysteine residue target of the S-glutathionylation reaction.
Materials and methods
Materials
The chromatographic columns MonoQTM 5/50 GL, Hi PrepTM Phenyl FF 16/10, SuperdexTM 75 10/30 HR and SuperdexTM 200 10/300 GL were from GE Healthcare. The Q-Sepharose chromatographic media, phenylmethanesulphonyl fluoride (PMSF), 5,5′-dithiobis-2-nitrobenzoic acid (DTNB), NADP+, NADPH, dithiothreitol (DTT), human insulin (10 mg mL−1 solution) and molecular weight protein standards for gel filtration were from Sigma-Aldrich. Electrophoretic materials were from Bio-Rad. All other chemicals were of analytical grade. Oligonucleotide synthesis and nucleotide sequencing were carried out by Primm (Italy).
The following buffers were used: buffer A, 20 mM Tris–HCl, pH 7.8; buffer B, 100 mM Tris–HCl, pH 7.8; buffer C, 100 mM potassium phosphate, pH 7.6, 10 mM EDTA; buffer D, 5 mM MgCl2, 10 % glycerol in buffer A; buffer PBS, phosphate buffer saline containing 10 mM sodium phosphate, pH 7.2 and 150 mM NaCl.
Purification of thioredoxin reductase and thioredoxin from Pseudoalteromonas haloplanktis
P. haloplanktis cells (50 g wet weight), grown in Luria–Bertani medium at 4 °C for 3 days (Masullo et al. 2000), were collected by centrifugation at 3000×g for 60 min at 4 °C and then resuspended in 150 mL buffer D supplemented with 1 mM PMSF. Cells were then disrupted by two passages through a Constant cell disruption system (Constant Systems Ltd., UK) at 1.5 MPa and the suspension was centrifuged at 30,000×g for 60 min at 4 °C to obtain the cell extract (S-30). This sample was ultra-centrifuged at 100,000×g for 2.5 h at 4 °C to obtain the soluble protein fraction (S-100). After extensive dialysis against buffer A, the S-100 was applied to a Q-Sepharose Fast-Flow column (1.6 × 60 cm) equilibrated with the same buffer. The column was washed with buffer A to remove unbound proteins; afterwards, bound proteins were eluted by a linear 0–450 mM NaCl gradient in buffer A (total volume 1200 mL) at a flow rate of 2 mL min−1. Fractions were checked for Trx and TrxR activity, which eluted at a different NaCl concentration; in particular, the endogenous TrxR and Trx from P. haloplanktis (PhTrxR and PhTrx, respectively) eluted at 250–280 mM and 350–400 mM NaCl, respectively. To evaluate protein purity, the active fractions were also analysed by SDS-PAGE.
In order to purify PhTrxR, fractions containing this activity were pooled together and dialysed against buffer A supplemented with 600 mM (NH4)2SO4 and then applied to a Hi PrepTM Phenyl FF 16/10 column, equilibrated with the same buffer. Bound proteins were eluted by a linear 600–20 mM (NH4)2SO4 inverse gradient in buffer A (total volume 480 mL) at a flow rate of 2 mL min−1. Active fractions were dialysed against buffer A supplemented with 150 mM NaCl and loaded onto a Mono QTM 5/50 GL, equilibrated with the same buffer. PhTrxR was eluted by a linear 150–500 mM NaCl gradient in buffer A (70 mL total volume) at a flow rate of 1 mL min−1.
For the purification of PhTrx, fractions from the Q-Sepharose chromatography containing this activity were pooled together, concentrated and then loaded onto a Superdex 200 10/300 GL column equilibrated with buffer A supplemented with 100 mM NaCl, at a flow rate of 0.75 mL min−1. The fractions displaying PhTrxR or PhTrx activity in the respective last chromatographic step, and showing a single protein band on SDS-PAGE, were pooled together, concentrated, dialysed against buffer A supplemented with 50 % glycerol and stored at −20 °C until use. Following this procedure, nearly 3 mg of pure PhTrxR and 2 mg of pure PhTrx from 50 g of wet cells were obtained.
Enzymatic assays
Enzymatic assays were carried out using a Cary 50 spectrophotometer (Varian), equipped with an electronic temperature controller. The activity of PhTrxR was determined by the DTNB reduction method (Luthman and Holmgren 1982). Briefly, the reaction mixture contained 5 mM DTNB and appropriate amounts of PhTrxR in 1 mL final volume buffer C. Unless otherwise indicated, the assay was carried out at 20 °C and started by the addition of 250 μM final concentration of NADPH. The reaction was followed kinetically by measuring the increase of the absorbance at 412 nm, due to the formation of TNB (εM = 13.6 mM−1 cm−1). Blanks run in the absence of PhTrxR were subtracted. One unit (U) of PhTrxR activity was defined as the amount of enzyme that caused the conversion of 1 μmol DTNB in 1 min. The specific activity of PhTrxR was expressed as U mg−1.
The activity of the PhTrx was determined by the insulin precipitation method (Holmgren 1979a). The standard assay mixture contained 0.13 mM human insulin and 0.5–15 μM PhTrx in 700 μL final volume buffer C. The reaction was carried out at 20 °C and began with the addition of DTT at 0.5 mM final concentration. The increase of absorbance, due to precipitation of the reduced β-chain of insulin, was monitored at 650 nm up to 60 min, to determine the rate of insulin reduction in the linear part of the increase. The activity of PhTrx was expressed as arbitrary units, corresponding to the value of ΔE 650 min−1.
In order to evaluate the combined activity of PhTrxR and PhTrx in the reconstituted thioredoxin system, 1μM PhTrxR was added to a reaction mixture containing 1–20 μM PhTrx and 0.13 mM human insulin in 700 μL final volume buffer C. The reaction started by the addition of NADPH at 200 μM final concentration and was followed at 20 °C monitoring the insulin precipitation at 650 nm and, when indicated, also the NADPH consumption at 340 nm (Holmgren 1979b; Spyrou et al. 1997); the values of ΔE 340 min−1 were measured in the linear range of the reaction, where no interference occurred by the insulin precipitation (ΔE 650 min−1).
The kinetic parameters of the PhTrxR reaction were derived at different temperatures from either the DTNB reduction or the insulin precipitation method in the reconstituted thioredoxin system. In the first assay the DTNB concentration ranged between 0.15 and 5 mM, whereas in the second one the PhTrx concentration varied between 0.5 and 15 μM; a fixed PhTrxR concentration of 0.12 or 1.03 μM was used in the first and second assay, respectively. The activity data were analysed by the Lineweaver–Burk method.
To determine the redox potential of the P. haloplanktis thioredoxin system, the reversibility of the NADPH-dependent reduction of PhTrx catalysed by PhTrxR in the absence of insulin was evaluated according to reaction 1. The assay was carried out at 25 °C as previously described (Krause et al. 1991; Jeon and Ishikawa 2002). In particular, the reaction mixture contained 60 μg PhTrx (in its initial PhTrx-S2 form) and 50 μM NADPH in 500 μL final volume buffer A. The value of the redox potential (E 0′(PhTrx)) was calculated according to the Nernst equation upon the sequential addition of 50 nM PhTrxR and 1.2 mM NADP+. The detailed procedure was previously reported (Cotugno et al. 2009). A value of −0.315 V was used for the \(E^{0\prime}_{({\text{NADP}}^{ + })}\) (Clark 1960).
Temperature-dependence studies on PhTrxR and PhTrx
The study of the temperature dependence of the reactions catalysed by PhTrxR and PhTrx, tested individually or in combination, was realised by determining the initial rate of the corresponding reactions from kinetics carried out at different temperatures, using the same protocol described in the previous paragraph. Data were analysed through the Arrhenius equation to obtain the activation energetic parameters of the reactions.
The thermal stability of PhTrxR was evaluated by measuring the residual DTNB reduction activity after the incubation of enzyme samples at different temperatures. The rate constants of heat inactivation (k in) were derived from a first order analysis of the residual activity. The k in values (s−1) were then analysed according to the Arrhenius equation to obtain the energetic parameters of the heat inactivation process.
Fluorescence-melting curves were obtained in the temperature interval 5–75 °C, using a computer-assisted Cary Eclipse Spectrofluorimeter (Varian) equipped with an electronic temperature controller. The increase in temperature was set at 0.2 °C min−1, and the emission in the aromatic region was monitored at every degree centigrade, using 280 and 345 nm for excitation and emission wavelength, respectively. Excitation and emission slits were both set at 10 nm and the samples were analysed in 500 μl stoppered cuvettes. Values of fluorescence intensity were subtracted for blanks run in the absence of the protein, corrected for temperature quenching, normalised between 0 and 100 %, and plotted versus temperature (Ruocco et al. 2004).
The heat stability of PhTrx was investigated through inactivation kinetics carried out at 95 °C. The residual PhTrx activity was determined at 20 °C by the insulin reduction method, using DTT as electron donor. The rate of insulin precipitation was compared to that measured on untreated PhTrx samples kept at 0 °C.
Parameters related to the amino acid composition of Trx and TrxR
The Trx and TrxR sequences were obtained (http://www.ncbi.nlm.nih.gov or http://www.expasy.ch) from microorganisms belonging to the eubacterial or archaeal kingdom and covering optimum growth temperatures from 7 to 103 °C; the list, including 42 microbial sources, was identical to that described in a previous work (De Vendittis et al. 2008). The number of proteins analysed (71 Trx and 45 TrxR) considered putative redundant isoforms, mainly for Trx; on the other hand, in some of the sources a canonic Trx or TrxR was apparently absent. The data of average mass and hydrophobicity per amino acid residue were obtained as previously reported (De Vendittis et al. 2008). The dependence of the average parameter on the growth temperature was evaluated as a linear curve fit of the data (y = a + b x) obtained with the least-squares method in which y represents the value of the average parameter, a the intercept at 0 °C, b the slope of the equation and x the optimum growth temperature of the source. The correlation of the data was estimated from the correlation coefficient r; the significance test included the calculation of the t Student parameter and the level was estimated by p.
Production of Cys → Ser mutants of PhTrxR
The production of a recombinant form of PhTrxR (rPhTrxR) through the expression vector vPhTrxR was previously described (Cotugno et al. 2009). Besides the two cysteine residues (C136 and C139) engaged in the disulphide bridge of its active site, PhTrxR possesses three other cysteines (C7, C106 and C303) as free thiols. In order to replace each of these residues with Ser, a site-directed mutagenesis was conducted on the vector vPhTrxR, using the following pairs of oligonucleotides, in which the base mismatch introduced to create the desired amino acid replacement is italicised. In particular, primers for C7S substitution were 5′-d-G7AA·GCA·AAA·CAT·AGT·AAG·TTA·CTT·ATT·TTA·GGC39-3′ and 5′-d-G39CC·TAA·AAT·AAG·TAA·CTT·ACT·ATG·TTT·TGC·TTC7-3′; primers for C106S substitution were 5′-d-G340GC·ACT·TAC·ACC·AGT·GAC·GCA·CTA·ATC·ATT·GC372-3′ and 5′-d-G372C·AAT·GAT·TAG·TGC·GTC·ACT·GGT·GTA·AGT·GCC340-3′; primers for C303S substitution were 5′-d-G892CC·GGA·ACA·GGT·AGT·ATG·GCA·GCA·TTA·GAT·GC923-3′ and 5′-d-G923C·ATC·TAA·TGC·TGC·CAT·ACT·ACC·TGT·TCC·GGC892-3′. The PCR, containing the vPhTrxR as template, the specific primers and the PfuTurbo™ DNA polymerase, was carried out according to the protocol of the QuikChange™ Site-Directed Mutagenesis Kit (Stratagene). The correct substitutions and the absence of other undesired mutations were confirmed by sequence analysis. After transformation of BL21(DE3) with the mutant plasmids, purification of the corresponding heterologous products, named C7S-PhTrxR, C106S-PhTrxR and C303S-PhTrxR, was obtained as described for rPhTrxR (Cotugno et al. 2009).
S-glutathionylation reaction
The reaction of S-glutathionylation was carried out as previously described (Castellano et al. 2008). Briefly, a 6.6-μM solution of PhTrxR or its recombinant and mutated forms in buffer A was incubated at room temperature for 1 h with increasing concentration of reduced glutathione (GSH), oxidised glutathione (GSSG) or S-nitrosoglutathione (GSNO) up to 10 mM. After incubation, the unbound glutathione forms were removed by ultrafiltration on Microcon-3 (Amicon) and the protein concentration of each sample was determined; aliquots were then run on SDS-PAGE under non reducing conditions. After SDS-PAGE, the protein samples were blotted onto an Immobilon P membrane (Millipore) and analysed by Western blotting using anti-glutathione monoclonal antibodies (Chemicon), as previously reported (Castellano et al. 2008).
Other methods
Protein purity was evaluated by SDS-PAGE according to standard protocols (Laemmli 1970). The molecular mass of PhTrxR and PhTrx under denaturing conditions was determined by SDS-PAGE on 12 and 15 % polyacrylamide gels, respectively. The molecular mass under non denaturing conditions was estimated by gel filtration on SuperdexTM 75 10/30 HR connected to a computer-assisted FPLC apparatus (GE Healthcare). The column was equilibrated at 4 °C with buffer A supplemented with 100 mM NaCl at a flow rate of 0.5 mL min−1. Protein concentration was determined by the method of Bradford (1976), using bovine serum albumin as standard. Fluorimetric measurements for the determination of the amount of FAD bound to PhTrxR were realised on a Cary Eclipse fluorescence spectrophotometer (Varian), according to the protocol previously described (Cotugno et al. 2009).
Results
Molecular properties of PhTrxR and PhTrx
The endogenous components of the thioredoxin system, PhTrxR and PhTrx, were isolated from a cell extract of P. haloplanktis. Under denaturing conditions, the molecular mass of PhTrxR and PhTrx was 34 and 12 kDa, respectively (not shown). These values are in good agreement with the theoretical values deduced from the amino acid sequence of PhTrxR (33.8 kDa) and PhTrx (11.9 kDa), respectively (Medigue et al. 2005). In order to get information on their structural organisation, the molecular mass of the psychrophilic proteins was also determined by gel filtration under non-denaturing conditions. Both PhTrxR and PhTrx eluted as single symmetric peaks and the molecular mass of PhTrxR was 68 kDa (not shown), thus clearly indicating that PhTrxR has a homodimeric organisation, as usually observed for other members of class II TrxR (Hirt et al. 2002; Windle et al. 2000); the molecular mass of PhTrx was 14 kDa, a value roughly similar to that obtained under denaturing conditions. Therefore, PhTrx should act as a monomeric protein, a feature typically observed for most of the Trx known to date (Jeon and Ishikawa 2002; Windle et al. 2000).
In the recombinant PhTrxR (rPhTrxR) it has been reported that a stoichiometric FAD/rPhTrxR ratio, ranging between 0.92 and 0.98, is essential for full activity of rPhTrxR (Cotugno et al. 2009). However, this ratio was reached only upon the addition of exogenous FAD. The amount of flavin cofactor bound to the endogenous PhTrxR was measured and the calculated molar ratio was 0.67. Although understoichiometric, the FAD/PhTrxR ratio was sufficient for evaluating the enzyme functionality; on the other hand, the flavin content reflected the FAD uptake during the P. haloplanktis growth. Therefore, the addition of exogenous FAD during purification of PhTrxR was not considered, to avoid a possible alteration of the functionality assay.
Functionality of the endogenous components of the thioredoxin system in P. haloplanktis
The functionality of PhTrxR and PhTrx was analysed in different assays, to assess that each endogenous component of the thioredoxin system from P. haloplanktis was purified in its active native form. The activity of PhTrxR was first measured with the synthetic substrate DTNB, using a reaction temperature of 20 °C, corresponding to the maximum growth temperature tolerated by P. haloplanktis. In particular, the kinetic parameters of the reduction reaction (k cat, 3 s−1; K M for DTNB, 2.6 mM), although not coincident with those previously reported for the recombinant PhTrxR (k cat, 6.9 s−1; K M for DTNB, 1.9 mM; Cotugno et al. 2009), indicate that the enzyme was purified in its active form. The difference could be explained with the understoichiometric FAD/PhTrxR ratio found in the endogenous flavoenzyme.
Concerning PhTrx, its activity was measured at 20 °C with the insulin precipitation method in the presence of DTT as electron donor. The rate of insulin reduction progressively increased upon addition of increasing amounts of PhTrx (Fig. 1); concomitantly, the lag phase in the insulin precipitation progressively decreased. Under these conditions, the apparent maximum rate of the reaction was 0.040 ΔE 650 min−1 and the PhTrx concentration leading to half maximal rate was 5 μM. These values indicate that also PhTrx was purified in its active form.

Thiol-disulphide oxidoreductase activity of PhTrx at 20 °C. The activity was measured with a continuous monitoring of the absorbance at 650 nm either in the absence (open circle) or in the presence of 0.9 μM PhTrx (filled circle), 2.2 μM PhTrx (filled triangle), 4.4 μM PhTrx (filled square), 8.9 μM PhTrx (filled diamond)
The availability of both endogenous components of the thioredoxin system of P. haloplanktis allowed an evaluation of their combined functionality in a reconstituted assay containing PhTrxR, PhTrx, NADPH as electron donor and human insulin as the thioredoxin substrate. The K M found for PhTrx was 2 μM, a value indicating the efficient interaction between the two thioredoxin components.
The full reversibility of the electron transfer between PhTrxR and PhTrx in the presence of NADPH/NADP+ was evaluated at 25 °C in the reconstituted thioredoxin system without insulin (not shown). The reaction was fully reversible and then associable to a cyclic process, thus confirming both the functionality and the composition of the thioredoxin system in P. haloplanktis. Moreover, this procedure allowed the evaluation of the redox potential of the PhTrx(SH)2/PhTrxS2 pair through the Nernst equation and the calculated E 0′(PhTrx) was −289 mV, a value indicating the reducing power of the psychrophilic thioredoxin system.
Effect of monovalent cations on the PhTrxR activity
The effect of chloride salts of selected monovalent cations on the DTNB reduction activity of PhTrxR was investigated. As shown in Fig. 2, all these salts were able to stimulate the activity of PhTrxR, although to a different extent. Among the salts used, NaCl at 0.5-M final concentration was the most effective, as it caused a fivefold increase in the activity measured in the absence of salt. The determination of the kinetic parameters of the DTNB reduction activity of PhTrxR in the presence of 0.5 M NaCl pointed to an improvement of both the affinity for the substrate and the catalytic constant of the reaction. Indeed the K M for DTNB was lowered to 1.3 mM, whereas the k cat increased to 7.6 s−1; therefore, a sixfold increase of the catalytic efficiency of PhTrxR was observed upon the addition of 0.5 M NaCl.

Effect of monovalent cations on the activity of PhTrxR. The DTNB reduction activity at 20 °C of 0.15 μ M PhTrxR was measured in the absence or in the presence of the indicated concentration of chloride salts of the following monovalent cations: Na+ (filled circle), K+ (open square), NH4 + (open triangle), Li+ (filled diamond) and Cs+ (open circle). The activity data were reported as a percentage of that measured in the presence of 0.5 M NaCl and represent mean values of three different determinations; the corresponding error bars are shown
Temperature dependence of PhTrxR and PhTrx activity
The temperature dependence of the reactions sustained by PhTrxR and PhTrx was investigated (Fig. 3). The activities promoted by the two isolated enzymes, namely the insulin precipitation by PhTrx (Fig. 3a) and the DTNB reduction by PhTrxR (Fig. 3b), were first evaluated. Both PhTrx and PhTrxR showed the maximum activity at around 30 °C, a value at least 15 °C higher than the optimal growth temperature of P. haloplanktis. The following decrease of activity was more evident in the DTNB reduction assay catalysed by PhTrxR. The temperature dependence of combined PhTrxR and PhTrx was also evaluated in the reconstituted system. This activity was followed by the insulin precipitation (Fig. 3c) and NADPH consumption (Fig. 3d). In the first case, the maximum activity shifted towards a higher temperature, around 40 °C, and remained almost constant up to 60 °C; only above this temperature, a significant lower activity was observed. In the second case, the fast NADPH reduction observed at temperatures higher than 20 °C impaired a correct determination of the activity above this temperature.

Effect of temperature on PhTrxR and PhTrx activities in different experimental conditions. a Activity of PhTrx measured through the insulin precipitation method. The reaction was carried out at the indicated temperatures and the final concentration of PhTrx was 10.1 μM. b Activity of PhTrxR measured through the DTNB reduction assay. The final concentration of PhTrxR was 0.014 μM. c Combined activity of PhTrx and PhTrxR measured through the insulin precipitation method. The final concentration of PhTrx and PhTrxR was 10.0 and 1.0 μM, respectively. d Same conditions as in c, with the exception that the activity was followed through the NADPH consumption. In all the experimental conditions the data were reported as mean values of three determinations with the corresponding error bars. Other details are indicated in “Materials and methods”
The studies on the effect of temperature included the determination of the energetic parameters of the enzymatic reactions promoted by PhTrx and PhTrxR. To this aim, the previously described data of activity were analysed according to the Arrhenius equation in the 5–20 °C temperature interval. As shown in Fig. 4, the linearity of the plots excluded temperature-dependent conformational changes in the range of temperature considered. This circumstance allowed the calculation of the energy of activation (E a) of the reactions. The lowest value of E a, 18.7 kJ mol−1, was measured with the reconstituted thioredoxin system, when the reaction was followed by the NADPH consumption; however, the E a raised to 47.1 kJ mol−1 when the insulin precipitation was used for activity measurements. Notably, a similar E a of 53.1 kJ mol−1 was obtained with the insulin precipitation method, when the activity of isolated PhTrx was evaluated. An intermediate value of 30.9 kJ mol−1 was calculated in the DTNB reduction assay. Therefore, the energetics of the various steps of the whole electron transfer from the initial donor NADPH to the final acceptor human insulin are affected by the interaction between PhTrxR and PhTrx and by the usage of a synthetic or heterologous electron acceptor/donor.

Arrhenius analysis of the activities sustained by PhTrxR and PhTrx in different experimental conditions. The data referred to the different activities reported in Fig. 3 were analysed according to the Arrhenius equation in the 5–20 °C temperature interval. Symbols as in Fig. 3. The squared correlation coefficient of data fitting ranged between 0.85 and 0.99
Effect of temperature on the stability of PhTrxR and PhTrx
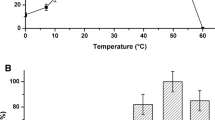
The thermostability of PhTrxR was investigated by heat inactivation studies and fluorescence-melting curves. In a first approach to evaluate the heat resistance of PhTrxR, a heat inactivation profile was determined from enzyme samples incubated for 10 min at temperatures ranging from 30 to 80 °C (Fig. 5a); the extrapolated half-inactivation temperature of PhTrxR was approximately 60 °C. The heat inactivation of PhTrxR was also followed kinetically and the process followed a first order kinetics at each temperature, thus allowing the calculation of the corresponding kinetic constants of heat inactivation (k in). The values of k in obtained in the 50–70 °C interval were analysed according to the Arrhenius equation (Fig. 5b) to obtain the energy of activation (E a) of the heat inactivation process, whose value was 154 kJ mol−1.

Heat stability of PhTrxR. a Heat inactivation profile. A 0.62-μM solution of PhTrxR in buffer A was incubated for 10 min at the indicated temperatures. The residual activity of the treated samples was determined after ice-chilling for 30 min, and expressed as a percentage of the untreated control kept at 0 °C. The data were reported as mean values of three determinations with the corresponding error bars. b Arrhenius plot of the heat inactivation process. Inactivation kinetics of PhTrxR were realised at temperatures ranging from 50 to 70 °C. To this aim, a 0.62-μM solution of the enzyme in buffer A was incubated at the indicated temperatures and, at selected times depending on the temperature chosen, an aliquot was withdrawn and assayed for the residual DTNB reduction activity. All kinetics were linear and the squared correlation coefficient of data fitting ranged between 0.87 and 0.99. The first-order k in values were used to draw the Arrhenius plot. c Fluorescence-melting curve of a 2-μM solution of PhTrxR in buffer A. Other details are indicated in “Materials and methods”
In the 5–75 °C interval the heat denaturation process of PhTrxR was also followed by fluorescence-melting curves, evaluating the increase of intrinsic fluorescence of the enzyme. The results reported in Fig. 5c indicate that the temperature for half denaturation of PhTrxR was approximately 55 °C, a value 5 °C lower than that determined through the heat inactivation profile. These data indicate that PhTrxR exhibits discrete heat resistance in spite of its psychrophilic origin.
Much more pronounced was the thermostability of the other component of the thioredoxin system in P. haloplanktis. This behaviour is clearly evident in the inactivation kinetics of PhTrx carried out at 95 °C and shown in Fig. 6. The calculated half-life of the psychrophilic protein was 263 min, a value pointing to the exceptional heat resistance of PhTrx in spite of its psychrophilic origin. The great heat resistance of PhTrx was also confirmed by the lack of any detectable effect in the fluorescence of this enzyme in the aromatic region of the spectrum, remaining constant between 20 and 99 °C (not shown).

Heat inactivation kinetics of PhTrx at 95 °C. A 16.5-μM PhTrx solution in buffer A was incubated at 95 °C. At the times indicated, aliquots were withdrawn and immediately chilled on ice for at least 30 min. The residual activity was measured through the insulin precipitation at a final PhTrx concentration of 2.8 μM. A t and A 0 represent the activities determined at the time t and zero, respectively. The squared correlation coefficient of data fitting was 0.98
Amino acid composition of PhTrx and PhTrxR and correlation with cold adaptation
The different temperature dependence and heat resistance of the two thioredoxin components prompted a phylogenetic analysis among the corresponding enzymes from other sources adapted at various growth temperatures in order to shed light on the behaviour of these psychrophilic enzymes. In particular, the evolutionary relationships were focused on the possible identification of specific structural requirements possessed by PhTrx and PhTrxR, which could explain their different cold adaptation. A previous study carried out on six model proteins covering various functional roles played by these macromolecules, showed that adaptation from cold to hot environments involved continuous and small adjustments of average parameters related to the amino acid composition (De Vendittis et al. 2008). Indeed, in all model proteins the average value per residue of mass and hydrophobicity almost linearly increased with the growth temperature of the host microorganism, even though some differences emerged in the “ideal proportion” of bulky and hydrophobic residues required for the adaptation to a selected temperature of each model protein. To this aim, parameters related to the amino acid composition of PhTrx and PhTrxR were compared to those of the corresponding enzymes from the same microorganisms analysed previously. As shown in Fig. 7, the average mass per residue in Trx (Fig. 7a) and TrxR (Fig. 7b) almost linearly increased with the growth temperature of the microorganism and a similar behaviour was also observed for the average hydrophobicity per residue of Trx (Fig. 7c) and TrxR (Fig. 7d). Therefore, the cold-adapted Trx and TrxR show a tendency towards smaller-sized and less hydrophobic residues in comparison to the mesophilic and the (hyper)thermophilic counterparts. However, significant differences emerged in the behaviour of the thioredoxin components, when considering the specific values of average mass and hydrophobicity of Trx or TrxR and the temperature-dependent increase of these parameters. The main difference resided in the significantly higher values of either average mass or hydrophobicity obtained for Trx with respect to TrxR. Moreover, at increasing temperatures, the distance between the average mass of Trx and TrxR slightly increased with temperature, whereas an opposite trend was observed for the average hydrophobicity. The extrapolated ideal values of average mass and hydrophobicity for the adaptation at 15 °C, the optimum growth temperature of P. haloplanktis, were 111.40 Da and 4.95 kJ mol−1 for Trx and 107.44 Da and 4.19 kJ mol−1 for TrxR. Interestingly, these figures are not far from the actual values calculated for PhTrx (109.87 Da and 5.15 kJ mol−1) and PhTrxR (106.92 Da and 4.10 kJ mol−1). The significant differences of these parameters between the two enzymes are likely related to their different cold adaptation and point to an increased usage of bulky and hydrophobic residues by PhTrx compared to PhTrxR.

Temperature dependence of average parameters related to the amino acid composition of Trx and TrxR form sources adapted from cold to hot environments. a Effect of growth temperature on the average mass per residue of Trx. Values of the linear fit: a = 110.89 Da; b = 0.03385 Da °C−1; r = 0.3644; t = 3.25; p < 0.005. b Effect of growth temperature on the average mass per residue of TrxR. Values of the linear fit: a = 107.12 Da; b = 0.02109 Da °C−1; r = 0.5432; t = 4.24; p < 0.001. c Effect of growth temperature on the average hydrophobicity per residue of Trx. Values of the linear fit: a = 4.90 kJ mol−1; b = 0.003722 kJ mol−1 °C−1; r = 0.4491; t = 4.17; p < 0.001. d Effect of growth temperature on the average hydrophobicity per residue of TrxR. Values of the linear fit: a = 4.06 kJ mol−1; b = 0.008703 kJ mol−1 °C−1; r = 0.8468; t = 10.44; p < 0.001. In each panel the filled circle indicates the actual value referred to the P. haloplanktis enzyme
S-glutathionylation of PhTrxR
Previous reports on superoxide dismutase from P. haloplanktis (PhSOD) proved that this enzyme has a highly reactive cysteine residue, forming disulphide adducts with either β-mercaptoethanol (Castellano et al. 2006) or cellular thiols, such as glutathione in its oxidised or nitrosylated form (Castellano et al. 2008). This covalent modification of PhSOD regulates the functions of this key anti-oxidant enzyme, thus suggesting that the S-glutathionylation reaction acts as a sensor of the cellular redox state of P. haloplanktis. Concerning PhTrxR, besides the two cysteines (C136 and C139) engaged in its active-site disulphide bridge, three other cysteines (C7, C103 and C303) are present as free thiols in the amino acid sequence of this flavoenzyme (Cotugno et al. 2009). These findings promoted an evaluation on the possible reactivity of PhTrxR towards glutathione. Therefore, PhTrxR was incubated with increasing concentrations of the oxidised form of glutathione (GSSG) and then analysed by Western blotting using anti-glutathione antibodies. As shown in Fig. 8a, the formation of a mixed disulphide between glutathione and PhTrxR was demonstrated by the presence of an immunoreactive band, which was already detectable in the sample treated with 0.1 mM GSSG; moreover, the immunoreactivity increased with the concentration of the modifying reagent. The glutathionylation by the nitrosylated (GSNO) or reduced (GSH) form of glutathione was also evaluated (not shown); however, immunoreactivity was evident starting from l mM GSNO or 10 mM GSH. These results suggest that, as already observed for PhSOD, PhTrxR is also the target of a S-glutathionylation reaction. The GSSG-treated samples were also assayed for DTNB reduction activity; however, the presence of the S-glutathionyl adduct did not apparently affect this catalytic property of PhTrxR (not shown).

S-glutathionylation of PhTrxR and its mutated forms. a Dose-dependent glutathionylation of endogenous PhTrxR. PhTrxR in buffer A was incubated at 25 °C in the absence (lane 1) or in the presence of 0.1 mM (lane 2), 1 mM (lane 3) or 10 mM (lane 4) GSSG. After removal of unbound glutathione, 0.9 μg of each protein sample was analysed by Western blotting. b Glutathionylation of recombinant mutated forms of PhTrxR. C7S-PhTrxR (lane 1, 2), C106S-PhTrxR (lane 3, 4), C303S-PhTrxR (lane 5, 6), or rPhTrxR (lane 7, 8) in buffer A was incubated at 25 °C in the absence (lane 1, 3, 5, 7) or in the presence of 10 mM GSSG (lane 2, 4, 6, 8). After removal of unbound glutathione, 0.9 μg of each protein sample was analysed by Western blotting. Other details are reported in “Materials and methods”
In order to identify the cysteine residue(s) target of the covalent modification by glutathione, three mutated forms of the flavoenzyme were obtained, in which each of the three free cysteines was replaced by a serine residue. The analysis by Western blotting of rPhTrxR and its mutated forms (C7S-rPhTrxR, C106S-rPhTrxR or C303S-rPhTrxR), untreated or treated with 1 mM GSSG, is shown in Fig. 8b. In the GSSG-treated samples, the immunoreactive band was present in rPhTrxR, C106S-rPhTrxR and C7S-rPhTrxR, although with different intensity; on the other hand, no reactivity at all was observed in the C303S-rPhTrxR sample. These data clearly suggest that C303 should be the target residue in the S-glutathionylation reaction of rPhTrxR.
Discussion
The molecular and functional properties of the purified endogenous components of the thioredoxin system isolated from the psychrophile Pseudoalteromonas haloplanktis have been investigated. The activity of this system, restoring the reduced state of cellular proteins after an oxidation reaction, is crucial for the survival of P. haloplanktis. Indeed, this microorganism is likely more exposed to the reactive oxygen species for both the increased oxygen solubility in the cold Antarctic sea and the enhanced stability of these toxic compounds at cold temperatures (Medigue et al. 2005; Pörtner et al. 2007). Previous work showed the different cold adaptation of the recombinant components of the thioredoxin system in P. haloplanktis (Cotugno et al. 2009). In this work, the biochemical characterisation of the purified endogenous PhTrx and PhTrxR confirmed the different temperature dependence and heat stability of the psychrophilic proteins and extended the study to the comparison of the energetic parameters of the reactions catalysed by these factors either alone or in combination. Moreover, the study included the effect of monovalent cations on the activity of PhTrxR, as well as the sensitivity of the flavoenzyme to the cellular thiol glutathione.
The study of the molecular properties of the purified endogenous components of the thioredoxin system in P. haloplanktis confirmed the previous data obtained on the recombinant counterparts (Cotugno et al. 2009). In particular, while PhTrxR is organised as a homodimer, PhTrx functions as a small monomeric protein, as usually found for most eubacterial components of the thioredoxin system. Furthermore, PhTrxR was purified as a flavoenzyme and the amount of FAD bound to the enzyme, although slightly understoichiometric, allowed the evaluation of the activity of the endogenous PhTrxR. Indeed, this FAD/PhTrxR ratio could reflect the required functionality of the enzyme, which activity is related to the FAD content. However, it cannot be excluded that the understoichiometric ratio was the consequence of the purification procedure.
PhTrx and PhTrxR were purified in their active oxidised form, as they catalysed the typical reactions sustained by these factors either alone or in combination. Indeed, when analysed alone, PhTrxR reduced the synthetic substrate DTNB using NADPH as electron donor, whereas PhTrx transferred the reducing equivalents from DTT to the heterologous substrate insulin. The kinetic parameters of these reactions were similar to those previously reported for the recombinant counterparts (Cotugno et al. 2009). When the combined activity of PhTrxR and PhTrx was analysed in a reconstituted thioredoxin system, the electron passage from the initial donor NADPH to the final acceptor insulin was demonstrated. Under these conditions, the K M of PhTrxR for its natural substrate PhTrx (2 μM) was similar to those reported for the mesophilic TrxR isolated from Escherichia coli (Ec) towards its two natural substrates, EcTrx1 (1.9 μM) and EcTrx2 (2.4 μM) (Miranda-Vizuete et al.1997). On the other hand, a higher K M (12.3 μM) was reported for the hyperthermophilic TrxR from Aeropyrum pernix (Ap) towards ApTrx (Jeon and Ishikawa 2002). Finally, the reversibility of the electron transfer in the reconstituted thioredoxin system allowed the determination of the E 0′(PhTrx), whose value (−289 mV) was similar to that recently reported for the recombinant system of P. haloplanktis (E 0′(rPhTrx) = −276 mV; Cotugno et al. 2009). Furthermore, no great differences were found with the corresponding parameters related to the mesophile E. coli (E 0′(EcTrx) = −270 mV; Krause et al. 1991) or the hyperthermophile A. pernix (E 0′(ApTrx) = −262 mV; Jeon and Ishikawa 2002). Therefore, all these data prove the interaction and functionality of the endogenous components of the thioredoxin system in P. haloplanktis.
The temperature dependence of the reactions associated to the electron transfer from the initial NADPH donor to the final acceptor human insulin was also investigated. In particular, a comparison was made among the values of E a found for each reduction step. The lowest E a (18.7 kJ mol−1) measured in the complete thioredoxin system, when the reaction was followed through the NADPH consumption, suggests that the first electron transfer from NADPH to the flavoenzyme is an energetically favoured process. Vice versa, when the activity of the complete thioredoxin system was followed through the insulin precipitation, the significant increase of E a (47.1 kJ mol−1) suggests that the final electron passage to the heterologous insulin could enhance the energetic barrier of the whole process. This suggestion is confirmed by the similar E a (53.1 kJ mol−1) obtained when the activity of PhTrx alone was evaluated through the insulin precipitation, using DTT as electron donor. The intermediate E a value (30.9 kJ mol−1) measured in the DTNB reduction assay catalysed by PhTrxR indicates that the energetic barrier of the electron passage from the flavoenzyme to the synthetic substrate DTNB, although higher than that measured in the first passage from NADPH to the flavoenzyme, remains lower than that obtained through the insulin precipitation assay. When a natural substrate for the psychrophilic PhTrx will be available, the redetermination of the E a value could be helpful for understanding the mechanism of the whole electron transfer, at least from the energetic point of view.
The studies on the thermostability of PhTrxR and PhTrx indicated that both psychrophilic proteins could tolerate temperatures significantly higher than those of the growth environment of P. haloplanktis. In the case of PhTrxR, the half-inactivation temperature of the flavoenzyme (60 °C) is well above the maximum growth temperature of 20 °C tolerated by P. haloplanktis. The energetics of the heat inactivation process of PhTrxR was also investigated and the calculated value of E a (154 kJ mol−1), although higher than that reported for polynucleotide phosphorylase (96.7 kJ mol−1), another enzyme isolated from P. haloplanktis (Evangelista et al. 2009), still reflects the psychrophilic origin of the source, as it is lower than the values usually reported for mesophilic and thermophilic enzymes, showing an average E a value of 278 ± 60 kJ mol−1 (Masullo et al. 1993; Grimaldi et al. 2008; Castellano et al. 2009; De Vendittis et al. 2010). Furthermore, since the denaturation profile of the flavoenzyme determined through fluorescence-melting curves is roughly similar to the heat inactivation profile, it is possible to conclude that heat inactivation and protein denaturation occur in the same temperature interval. These data, in agreement with the previous results obtained with the recombinant flavoenzyme (Cotugno et al. 2009) point to the discrete heat stability of PhTrxR.
In the case of PhTrx, the exceptional half-life measured in the inactivation kinetics at 95 °C (263 min) ranks this protein as the most thermostable from a psychrophilic origin. To our knowledge, the enzymes isolated from P. haloplanktis display a higher heat resistance compared to other psychrophiles. However, the half-life of other P. haloplanktis enzymes is significantly lower compared to PhTrx. Indeed, besides PhTrxR (half-life 10 min at 60 °C, this work), superoxide dismutase, another antioxidant enzyme (Castellano et al. 2006), and the elongation factor Ts (Raimo et al. 2004) display a lower heat resistance (half-life 10 min at 54 and 57 °C, respectively). Interestingly, PhTrx possesses a greater resistance to heat inactivation even in comparison to the mesophilic recombinant EcTrx2; in fact, 40 % of the insulin-reducing activity of EcTrx2 was lost after 5 min of incubation at 85 °C (Miranda-Vizuete et al. 1997). Moreover, the heat resistance of PhTrx is not very distant from that of Trx from a hyperthermophilic microorganism. For instance, at 92 °C the half-life of recombinant Trx-A1 and Trx-A2 from Sulfolobus solfataricus was 20.5 and 56.6 h, respectively (Grimaldi et al. 2008).
The analysis of some parameters related to the amino acid composition of PhTrx and PhTrxR gave a possible explanation on the different thermodependence and thermostability of the two psychrophilic thioredoxin components. Indeed, the significantly higher proportion of bulky and hydrophobic residues possessed by PhTrx made this protein intrinsically more compact compared to PhTrxR, a finding suggesting a structural basis for the exceptional thermostability of PhTrx. On the other hand, the consequent lower flexibility of PhTrx was not dramatic for its cold adaptation, probably because of its small size and the absence of large conformational changes during catalysis. Concerning PhTrxR, its lower proportion of bulky and hydrophobic residues ensured an appropriate flexibility of the flavoenzyme, which underwent a large conformational change during catalysis (Lennon et al. 2000), although reducing its thermostability compared to PhTrx.
The biochemical characterisation of PhTrxR included the effect of some monovalent cations on the DTNB reduction reaction catalysed by the flavoenzyme. The results showed that all monovalent cations enhance the activity of PhTrxR and that the highest stimulation, nearly sixfold increase, was reached upon the addition of 0.5 M NaCl. Therefore, PhTrxR could be classified as a polyextremophilic protein. A similar behaviour was already observed for other enzymes isolated from P. haloplanktis (Srimathi et al. 2007; Evangelista et al. 2009). Furthermore, the stimulation of the activity by monovalent cations is common to other psychrophilic proteins, thus indicating a possible common regulatory role exerted by these cations on the mechanism of action in psychrophiles.
Another interesting feature emerging from this work concerns the reactivity of PhTrxR towards the cellular thiol glutathione. In particular, the flavoenzyme was S-glutathionylated by the oxidised form of glutathione in a dose-dependent way; a reactivity was also observed with the nitrosylated and even the reduced form of this thiol, even though at tenfold and 100-fold higher doses, respectively. A similar behaviour was already observed for the PhSOD and in that case the covalent modification regulated the functions of the anti-oxidant enzyme (Castellano et al. 2008). Vice versa, in the case of PhTrxR the S-glutathionylation of the enzyme did not apparently cause a significant alteration of the DTNB reduction activity. In any case, the S-glutathionylation reaction must play a key role in controlling the cellular redox state of P. haloplanktis, because it was demonstrated that this covalent modification is induced by cellular oxidative stress conditions (Castellano et al. 2008). The mutagenic analysis of the three free cysteines possessed by PhTrxR allowed the identification of C303 as the target residue of the S-glutathionylation reaction on the basis of the lack of reactivity towards glutathione upon the C303S replacement. Indeed, the S-glutathionylation reaction was evident, even though to a different extent, with either C7S or C106S replacement. A clear explanation for the significant lower reactivity observed with the C7S-rPhTrxR mutant compared to wild type is unavailable at moment; when the 3D structure of the enzyme will be available, it will be interesting to evaluate if the C7S amino acid replacement could reduce the accessibility of the C303 residue of PhTrxR to the modification reaction by glutathione. However, on the basis of a 3D model, the residue C303, target of the covalent modification by glutathione, appears more exposed to solvent than other non catalytic cysteines.
Conclusions
The results reported in this paper indicate that the efficiency of the thioredoxin system is crucial for the preservation of the reduced state of cellular proteins in P. haloplanktis, even considering the possible involvement of glutathione in the alteration of the redox state of the free cysteine residues possessed by the cellular proteins of this psychrophile and the activity of components of another thiol-disulphide oxidoreductase system aimed at the formation of disulphide bonds (PhDsbA and PhDsbA2) in the periplasmic space (Madonna et al. 2006). The most relevant finding of this work was the high thermal stability of both components of the thioredoxin system, mainly for PhTrx. It is likely that the exceptional heat resistance of the thioredoxin system in P. haloplanktis reflects an adaptation mechanism of this psychrophile, considering its growth under oxidative stress conditions caused by the increased oxygen solubility in the cold environment.
Abbreviations
- Ap:
-
Aeropyrum pernix
- Ec :
-
Escherichia coli
- Ph :
-
Pseudoalteromonas haloplanktis
- DTNB:
-
5,5′-dithiobis-2-nitrobenzoic acid
- DTT:
-
Dithiothreitol
- GSH:
-
Reduced glutathione
- GSSG:
-
Oxidised glutathione
- GSNO:
-
Nitrosylated glutathione
- PMSF:
-
Phenylmethanesulphonyl fluoride
- TNB:
-
2-nitro-5-thiobenzoate
- Trx:
-
Thioredoxin
- TrxR:
-
Thioredoxin reductase
- Trx-S2 and Trx-(SH)2 :
-
Oxidised and reduced form of Trx, respectively
- k in :
-
Heat inactivation rate constant
References
Arnér ESJ, Holmgren A (2000) Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem 267:6102–6109
Arscott LD, Gromer S, Schirmer RH, Becker K, Williams CH Jr (1997) The mechanism of thioredoxin reductase from human placenta is similar to the mechanisms of lipoamide dehydrogenase and glutathione reductase and is distinct from the mechanism of thioredoxin reductase from Escherichia coli. Proc Natl Acad Sci 94:3621–3626
Birolo L, Tutino ML, Fontanella B, Gerday C, Mainolfi K, Pascarella S, Sannia G, Vinci F, Marino G (2000) Aspartate aminotransferase from the Antarctic bacterium Pseudoalteromonas haloplanktis TAC 125. Cloning, expression, properties, and molecular modelling. Eur J Biochem 267:2790–2802
Bradford M (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Castellano I, Di Maro A, Ruocco MR, Chambery A, Parente A, Di Martino MT, Parlato G, Masullo M, De Vendittis E (2006) Psychrophilic superoxide dismutase from Pseudoalteromonas haloplanktis: biochemical characterization and identification of a highly reactive cysteine residue. Biochimie 88:1377–1389
Castellano I, Ruocco MR, Cecere F, Di Maro A, Chambery A, Michniewicz A, Parlato G, Masullo M, De Vendittis E (2008) Glutathionylation of the iron superoxide dismutase from the psychrophilic eubacterium Pseudoalteromonas haloplanktis. Biochim Biophys Acta 1784:816–826
Castellano I, Cecere F, De Vendittis A, Cotugno R, Chambery A, Di Maro A, Michniewicz A, Parlato G, Masullo M, Avvedimento EV, De Vendittis E, Ruocco MR (2009) Rat mitochondrial manganese superoxide dismutase: amino acid positions involved in covalent modifications, activity, and heat stability. Biopolymers 91:1215–1226
Chae HZ, Chung SJ, Rhee SG (1994) Thioredoxin-dependent peroxide reductase from yeast. J Biol Chem 269:27670–27678
Clark WM (1960) Oxidation-reduction potentials of organic systems. The Williams & Wilkins Co., Baltimore
Cotugno R, Ruocco MR, Marco S, Falasca P, Evangelista G, Raimo G, Chambery A, Di Maro A, Masullo M, De Vendittis E (2009) Differential cold-adaptation among protein components of the thioredoxin system in the psychrophilic eubacterium Pseudoalteromonas haloplanktis TAC 125. Mol BioSyst 5:519–528
De Vendittis E, Castellano I, Cotugno R, Ruocco MR, Raimo G, Masullo M (2008) Adaptation of model proteins from cold to hot environments involves continuous and small adjustments of average parameters related to amino acid composition. J Theor Biol 250:156–171
De Vendittis A, Amato M, Mickniewicz A, Parlato G, De Angelis A, Castellano I, Rullo R, Riccitiello F, Rengo S, Masullo M, De Vendittis E (2010) Regulation of the properties of superoxide dismutase from the dental pathogenic microorganism Streptococcus mutans by iron- and manganese-bound co-factor. Mol BioSyst 6:1973–1982
Ejiri SI, Weissbach H, Brot N (1979) Reduction of methionine sulfoxide to methionine by Escherichia coli. J Bacteriol 139:161–164
Evangelista G, Falasca P, Ruggiero I, Masullo M, Raimo G (2009) Molecular and functional characterization of polynucleotide phosphorylase from the Antarctic eubacterium Pseudoalteromonas haloplanktis. Protein Pept Lett 16:999–1005
Gasdaska PY, Berggren MM, Berry MJ, Powis G (1999) Cloning, sequencing and functional expression of a novel human thioredoxin reductase. FEBS Lett 442:105–111
Ghisla S, Massey V (1989) Mechanisms of flavoprotein-catalyzed reactions. Eur J Biochem 181:1–17
Gilbert HF (1990) Molecular and cellular aspects of thiol-disulfide exchange. Adv Enzymol Relat Areas Mol Biol 63:69–172
Grimaldi P, Ruocco MR, Lanzotti MA, Ruggiero A, Ruggiero I, Arcari P, Vitagliano L, Masullo M (2008) Characterisation of the components of the thioredoxin system in the archaeon Sulfolobus solfataricus. Extremophiles 12:553–562
Hernandez HH, Jaquez OA, Hamill LJ, Elliot SJ, Drennan CL (2008) Thioredoxin reductase from Thermoplasma acidophilum: a new twist on redox regulation. Biochemistry 47:9728–9737
Hirt RP, Müller S, Embley TM, Coombs GH (2002) The diversity and evolution of thioredoxin reductase: new perspectives. Trends Parasitol 18:302–308
Holmgren A (1979a) Thioredoxin catalyzes the reduction of insulin disulfides by dithiothreitol and dihydrolipoamide. J Biol Chem 254:9627–9632
Holmgren A (1979b) Reduction of disulfides by thioredoxin. Exceptional reactivity of insulin and suggested functions of thioredoxin in mechanism of hormone action. J Biol Chem 254:9113–9119
Holmgren A (1985) Thioredoxin. Annu Rev Biochem 54:237–271
Holmgren A (1989) Thioredoxin and glutaredoxin systems. J Biol Chem 264:13963–13966
Huber HE, Tabor S, Richardson CC (1987) Escherichia coli thioredoxin stabilizes complexes of bacteriophage T7 DNA polymerase and primed templates. J Biol Chem 262:16224–16232
Jeon SJ, Ishikawa K (2002) Identification and characterization of thioredoxin and thioredoxin reductase from Aeropyrum pernix K1. Eur J Biochem 269:5423–5430
Kanzok SM, Fechner A, Bauer H, Ulschmid JK, Müller HM, Botella-Munoz J, Schneuwly S, Schirmer R, Becker K (2001) Substitution of the thioredoxin system for glutathione reductase in Drosophila melanogaster. Science 291:643–646
Kashima Y, Ishikawa K (2003) A hyperthermostable novel protein-disulfide oxidoreductase is reduced by thioredoxin reductase from hyperthermophilic archaeon Pyrococcus horikoshii. Arch Biochem Biophys 418:179–185
Kern R, Malki A, Holmgren A, Richarme G (2003) Chaperone properties of Escherichia coli thioredoxin and thioredoxin reductase. Biochem J 371:965–972
Krause G, Lundstrom J, Barea JL, Pueyo de la Cuesta C, Holmgren A (1991) Mimicking the active site of protein disulfide isomerase by substitution of proline 34 in Escherichia coli thioredoxin. J Biol Chem 266:9494–9500
Ladenstein R, Ren B (2006) Protein disulfides and protein disulfide oxidoreductases in hyperthermophiles. FEBS J 273:4170–4185
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lennon BW, Williams CH Jr, Ludwig ML (2000) Twists in catalysis: alternating conformations of Escherichia coli thioredoxin reductase. Science 289:1190–1194
Luthman M, Holmgren A (1982) Rat liver thioredoxin and thioredoxin reductase: purification and characterization. Biochemistry 21:6628–6633
Madonna S, Papa R, Birolo L, Autore F, Doti N, Marino G, Quemeneur E, Sannia G, Tutino ML, Duilio A (2006) The thiol-disulfide oxidoreductase system in the cold-adapted bacterium Pseudoalteromonas haloplanktis TAC 125: discovery of a novel disulfide oxidoreductase enzyme. Extremophiles 10:41–51
Masullo M, Raimo G, Bocchini V (1993) Resistance of archaebacterial aEF-1alpha·GDP against denaturation by heat and urea. Biochim Biophys Acta 1162:35–39
Masullo M, Arcari P, de Paola B, Parmeggiani A, Bocchini V (2000) Psychrophilic elongation factor Tu from the Antarctic Moraxella s. Tac II25: biochemical characterization and cloning of the encoding gene. Biochemistry 39:15531–15539
Matthews JR, Wakasugi N, Virelizier JL, Yodoi J, Hay RT (1992) Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res 20:3821–3830
Medigue C, Krin E, Pascal G, Barbe V, Bernsel A, Bertin PN, Cheung F, Cruveiller S, D’Amico S, Duilio A, Fang G, Feller G, Ho C, Mangenot S, Marino G, Nilsson J, Parrilli E, Rocha EPC, Rouy Z, Sekowska A, Tutino ML, Vallenet D, von Heijne G, Danchin A (2005) Coping with cold: the genome of the versatile marine Antarctica bacterium Pseudoalteromonas haloplanktis TAC125. Genome Res 15:1325–1335
Miranda-Vizuete A, Damdimopoulos AE, Gustafsson J, Spyrou G (1997) Cloning, expression, and characterization of a novel Escherichia coli thioredoxin. J Biol Chem 272:30841–30847
Moore EC, Reichard P, Thelander L (1964) Enzymatic synthesis of deoxyribonucleotides. V. Purification and properties of thioredoxin reductase from Escherichia coli B. J Biol Chem 239:3445–3452
Pörtner HO, Peck L, Somero G (2007) Thermal limits and adaptation in marine Antarctic ectotherms: and integrative view. Phil Trans R Soc B 362:2233–2258
Raimo G, Lombardo B, Masullo M, Lamberti A, Longo O, Arcari P (2004) Elongation factor Ts from the Antarctic eubacterium Pseudoalteromonas haloplanktis TAC 125: biochemical characterization and cloning of the encoding gene. Biochemistry 43:14759–14766
Ruocco MR, Ruggiero A, Masullo L, Arcari P, Masullo M (2004) A 35 kDa NAD(P)H oxidase previously isolated from the archaeon Sulfolobus solfataricus is instead a thioredoxin reductase. Biochimie 86:883–892
Schenk H, Klein M, Erdbrügger W, Dröge W, Schulze-Osthoff K (1994) Distinct effects of thioredoxin and antioxidants on the activation of transcription factors NF-kappa B and AP-1. Proc Natl Acad Sci USA 91:1672–1676
Spyrou G, Enmark E, Miranda-Vizuete A, Gustaffson J (1997) Cloning and expression of a novel mammalian thioredoxin. J Biol Chem 272:2936–2941
Srimathi S, Jayaraman G, Feller G, Danielsson B, Narayanan PR (2007) Intrinsic halotolerance of the psychrophilic α-amylase from Pseudoalteromonas haloplanktis. Extremophiles 11:505–515
Tsang ML, Schiff JA (1976) Sulfate-reducing pathway in Escherichia coli involving bound intermediates. J Bacteriol 125:923–933
Williams CH Jr (1995) Mechanism and structure of thioredoxin reductase from Escherichia coli. FASEB J 9:1267–1276
Windle HJ, Fox A, Ni Eidhin D, Kelleher D (2000) The thioredoxin system of Helicobacter pylori. J Biol Chem 275:5081–5089
Acknowledgments
This work was supported by grants from MIUR, PRIN 2009 (Rome) awarded to MM, EDV, GR.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by F. Robb.
Authors P. Falasca and G. Evangelista equally contributed to this work.
Rights and permissions
About this article
Cite this article
Falasca, P., Evangelista, G., Cotugno, R. et al. Properties of the endogenous components of the thioredoxin system in the psychrophilic eubacterium Pseudoalteromonas haloplanktis TAC 125. Extremophiles 16, 539–552 (2012). https://doi.org/10.1007/s00792-012-0453-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-012-0453-0