
Abstract
Sulfur oxygenase reductase (SOR) enzyme is responsible for the initial oxidation step of elemental sulfur in archaea. Curiously, Aquifex aeolicus, a hyperthermophilic, chemolithoautotrophic and microaerophilic bacterium, has the SOR-encoding gene in its genome. We showed, for the first time the presence of the SOR enzyme in A. aeolicus, its gene was cloned and recombinantly expressed in Escherichia coli and the protein was purified and characterised. It is a 16 homo-oligomer of approximately 600 kDa that contains iron atoms indispensable for the enzyme activity. The optimal temperature of SOR activity is 80°C and it is inactive at 20°C. Studies of the factors involved in getting the fully active molecule at high temperature show clearly that (1) incubation at high temperature induces more homogeneous form of the enzyme, (2) conformational changes observed at high temperature are required to get the fully active molecule and (3) acquisition of an active conformation induced by the temperature seems to be more important than the subunit number. Differences between A. aeolicus SOR and the archaea SORs are described.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Oxidation and reduction of sulfur compounds are vital processes for many prokaryotes, and essential steps in the global sulfur cycle. Elemental sulfur and sulfur compounds are the most abundant sources of both acceptors and donors in volcanic environments and are used by many ancestral microorganisms to support growth (Kletzin et al. 2004). However, because of the multiple oxidation states, the biochemistry and chemistry of the sulfur cycle are complex and not completely understood (Suzuki 1999; Hedderich et al. 1998).
In the past it was postulated that different pathways for sulfur oxidation occurred in prokaryotes. The characterisation of the sulfur oxidising enzyme system (SOX) of the alphaproteobacterium Paracoccus pantotrophus (Rother et al. 2001) and the identification of the respective genes in other chemotrophic or phototrophic bacteria raised the question of “emergence of a common mechanism” in Bacteria (Friedrich et al. 2005) also referred as thiosulfate oxidising multienzyme system (TOMES). Up to date, the SOX system has been only found in the members of the domain Bacteria.
Archaea use different pathways to oxidise sulfur compounds. The best studied sulfur-oxidising enzymes is a soluble, cytoplasmic sulfur oxygenase reductase (SOR) (Kletzin et al. 2004) from the facultative anaerobic, chemolithoautotrophic, thermoacidophilic archaeon Acidianus ambivalens. The SOR catalyses an oxygen-dependent sulfur disproportionation reaction leading to sulfite and sulfide as reaction products (Kletzin 1989; Urich et al. 2004). The catalytic reaction can be split into a sulfur oxygenation reaction S + O2 + H2O → HSO3 − + H+ and a sulfur disproportionation reaction 3S + 3H2O → HSO3 − + 2HS− + 3H+. The sum of these reactions gives 4S + O2 + 4H2O → 2HSO3 − + 2HS− + 4H+. A nonenzymic reaction leading to the formation of thiosulfate is also observed under excess of S°:S + HSO3 − → S2O3 2− + H+. The X-ray structure of the SOR from A. ambivalens has been determined recently (Urich et al. 2006). It is a 24-subunit enzyme of 871 kDa (24 identical subunits) structurally similar to the ferritins. A cysteine persulfide and a low reduction potential mononuclear non-heme iron site ligated by a 2-His-1-carboxylate facial triad in a pocket of each subunit constitute the active sites of the enzyme. Considering its cytoplasmic location, the SOR does not contribute to the formation of a transmembrane proton gradient and it probably provides soluble precursors like thiosulfate for membrane-bound enzymes such as thiosulfate:quinone oxidoreductase (TQO) (Müller et al. 2004). Up to date, only characterisations of archaeal SOR have been published in the literature. Aquifex aeolicus (VF5) is a hyperthermophilic, microaerophilic and chemolithoautotrophic bacterium capable of growing under H2/CO2/O2 atmosphere in a medium containing only inorganic compounds (Huber et al. 1992). Phylogenetic analyses based on 16S rRNA sequence comparisons showed that Aquificales represents the earliest branch in the bacterial domain (Nübel et al. 2000). The complete genome sequence of A. aeolicus has been published (Deckert et al. 1998). Numerous genes encoding proteins involved in the sulfur metabolism have been detected and a metabolic pathway from hydrogen oxidation to sulfur reduction has been identified (Guiral et al. 2005). Genes encoding proteins involved in sulfur oxidation pathway are also detected. Among these, genes encoding proteins homologous to bacterial SOX proteins and to the archaeal SOR enzyme have been identified. The presence of a putative sor gene in the hyperthermophilic bacterium A. aeolicus led to consider about the biochemical and biophysical properties of this bacterial enzyme and its function in this organism.
Here, we report the presence of SOR homolog in the bacterial organism, A. aeolicus. Its gene has been cloned and recombinantly expressed in E. coli. For the first time, the characterisation of a bacterial SOR is described. The role of the temperature in the acquisition of the active form of the recombinant and native SORs have been studied in details.
Materials and methods
All restriction enzymes were obtained from Promega. PCR was carried out using DNA polymerase (Pwo) from Roche Applied Sciences. DNA ligase was obtained from Roche Applied Sciences. Cloning oligonucleotides were purchased from MWG and DNA sequencing was performed by Genome Express.
Strains and media
Growth of E. coli BL21-CodonPlus(DE3)-RIPL strains (Stratagene) was carried out in LB medium (Maniatis et al. 1982), supplemented with the appropriate antibiotic at a final concentration of 0.27 mM for ampicillin and 0.15 mM for chloramphenicol when specified.
Aquifex aeolicus VF5 was grown as described by Stetter (1983). The cells were harvested in the late exponential growth phase.
Cloning of the sor gene
The ORF aq_455 encoding SOR was amplified from A. aeolicus genomic DNA by two PCR reactions using, respectively, oligonucleotides sor1-left (5′-tatgctgacagatattaaaaaagg-3′) and sor1-right (5′-cttaaggaaagagaatcagatcc-3′) for the first PCR and oligonucleotides sor2-left (5′-tgctgacagatattaaaaaagg-3′) and sor2-right (5′tggacttaaggaaagagaatcagatcc-3′) for the second PCR. The two PCR products were mixed, denatured during 2 min at 90°C and matched by a linear gradient from 90 to 25°C (0.1°C/min). The mix was ligated into pET22b(+) (Novagen) double digested by NdeI–SalI to obtain pETSOR. The sequence of the insert of 1,000 pb in the resulting plasmid was verified by DNA sequencing.
Protein overproduction and purification
Escherichia coli BL21-CodonPlus(DE3)-RIPL was transformed with the pETSOR plasmid. Culture (10 l) of recombinant E. coli was grown at 37°C until OD600 of 1 and then induced with 1 mM isopropyl β-d-thiogalactopyranoside (IPTG) for 3 h at 37°C. Cells were harvested by centrifugation and were disrupted by French press in buffer A (20 mM Tris–HCl, pH 7.4 containing 100 mM NaCl, 10% glycerol) containing proteases inhibitors cocktail from Roche Applied Science. The crude extract was centrifuged at 14,000g for 10 min and the supernatant heated at 80°C for 40 min to precipitate heat-labile E. coli proteins. Following a centrifugation at 4,000g for 15 min, the supernatant containing the overproduced protein was concentrated using Centriprep concentrators (Amicon) with YM-30 membranes and was loaded onto a Superdex S200 high resolution column (FPLC apparatus, Amersham Pharmacia Biotech) equilibrated and eluted in the buffer A. Active fractions were concentrated, frozen in liquid nitrogen and stored at −20°C.
Wild-Type SOR Purification
Aquifex aeolicus cells (40 g) were resuspended in buffer B (20 mM Tris–HCl, pH 7.4, 5 mM EDTA), disrupted by a French press and centrifuged at 60,000g for 45 min. The soluble extract containing the SOR activity was applied onto a CMC column equilibrated in buffer B. Fractions with SOR activity were loaded onto a Q-Sepharose column (FPLC apparatus, Amersham Pharmacia Biotech) equilibrated with 20 mM Tris–HCl, pH 7.4. Proteins were eluted by a gradient of NaCl from 0 to 500 mM. Active fractions were concentrated on centricon YM-30 and loaded onto a Superdex S200 equilibrated with 20 mM Tris–HCl, pH 7.6, 100 mM NaCl. Active fractions were concentrated, frozen in liquid nitrogen and stored at −20°C.
Enzyme activities
The SOR activity was assayed by following the evolution of hydrogen sulfide, sulfite and thiosulfate. The assay was performed by incubating the enzyme for 40 min with 5% flowers of sulfur in 50 mM Tris–HCl pH 7.4 at 85°C. H2S concentration was determined as described by Guiral et al. (2005). As this method does not take account the loss of sulfide through oxidation, the activities determined are underestimated. Sulfite and thiosulfate concentrations were determined by the method described by (Kletzin 1989). One unit of SOR activity corresponds to the uptake of 1 μmol of hydrogen sulfide (reductase reaction) and 1 μmol of thiosulfate plus sulfite (oxygenase reaction) formed per minute. Specific activities were expressed as units of sulfite plus thiosulfate formed per milligram of proteins or units of hydrogen sulfide formed per milligram of protein.
The pH studies were performed by using 100 mM sodium acetate from pH 3.5 to 5.5, 100 mM MES from pH 5.5 to 6.5, 100 mM MOPS from pH 6.5 to 7.5 and 100 mM Tris–HCl from pH 7.5 to 9; 5 μg of SOR were used to determine the enzyme activity at each pH.
SOR activity on non-denaturing gels was detected by the consumption of polysulfide substrate; 2 mM polysulfide was generated by addition of 2 mM of Na2S4O6–8 mM Na2S in 50 mM Tris–HCl pH 8.3. SOR activity was visualised by adding 2,3,5-triphenyltetrazolium chloride to the gel pre-incubated 60 min at 80°C in 50 mM Tris–HCl pH 7.4 in the presence of polysulfide. Polysulfide consumption by SOR is revealed by the presence of a white band.
N-terminal sequence determination
The N-terminal amino acid sequence of the SOR was determined with an Applied Biosystems Procise 494 microsequencer. Quantitative determination of phenylthiohydantoin derivates was done by high-pressure liquid chromatography (Water Associates, Inc.) monitored by a data and chromatography control station (Waters).
Denaturing gel electrophoresis
Thirty milligrams of recombinant SOR were loaded on a 4% polyacrylamide stacking/10% running SDS gel (Mini-protean II, Bio-Rad).
Native gel electrophoresis and immunoblotting
Ten milligrams of soluble extract from A. aeolicus cells and 35 ng of recombinant SOR were loaded on a 4% polyacrylamide stacking, 10% running gel (Mini-Protean II; Bio-Rad). After migration, Western blotting was performed using standard procedures. Anti-SOR antibodies against A. aeolicus recombinant SOR were used and the detection reaction was performed using goat peroxidase conjugated anti-rabbit IgG (Sigma) and SuperSignal West Pico Chemiluminescent Substrat reagents (Pierce).
Blue-native gels (BN gels)
BN-PAGE is a charge shift method in which the electrophoretic mobility is determined by the negative charge of the bound coomassie dye and the size and shape of the protein. BN gels were performed using a mini-Protean II cell electrophoresis apparatus (Bio-Rad) according to the method of Schägger (Schägger and Von Jagow 1991; Schägger et al. 1994). Wild-type or recombinant proteins were supplemented with 2 μl of a 0.5% stock solution of coomassie blue G250 (Serva Blue G, Serva Electrophoresis, Heidelberg) in 500 mM aminocaproïc acid. Samples were loaded on 5–15% gradient gels with Native Mark Protein Standard (Invitrogen). After electrophoresis, the gel was scanned in a GS 800 densitometer from Bio-Rad. The results were analyzed with Quantity One software (Bio-Rad).
Protein identification by mass spectrometry and database search
Tryptic digestion of excised gel plugs was described previously (Brugna-Guiral et al. 2003). Protein identification was performed by peptide mass finger printing on a MALDI-TOF MS mass spectrometer DE-RP (Applied Biosystems) exploiting the non-redundant database NCBInr (NIH, Bettesda, USA) using MASCOT search (http://www.matrixscience.com).
Circular dichroism
CD spectra were recorded on a Jasco J-715 spectropolarimeter equipped with a Peltier-type temperature control system (model PTC-348WI) between 195 and 250 nm for the far-UV and 250 and 340 nm for the near-UV. Experiments conditions were 0.2 nm/min; temperatures were 20 and 80°C. Spectra were averaged from five acquisitions. Protein concentration was 0.05 mg/ml in 20 mM Tris–HCl, pH 7.6 buffer. The CD spectra were analysed with the CD Spectroscopy Deconvolution CDNN 2.1 software for determining the secondary-structure classes and K2d algorithm (Morris et al. 1999). To determine the effect of aromatic residues on SOR secondary structures, CD spectra were also monitored between 250 and 340 nm from 20 to 90°C (1°C/min). The experimental data, i.e, plot of the observed ellipticity against temperature, were fitted with the data analysis program Sigma plot to the equation reported by Tan et al. (1995).
Sequence analysis
Sequences were aligned using ClustalW (Thompson et al. 1994) at http://pbil.univ-lyon1.fr/ and consensus secondary structure predictions were made using http://www.expasy.org and http://npsa-pbil.ibcp.fr/ (Guermeur et al. 1999).
Results
Sequence analysis of the sor gene
Analysis of the A. aeolicus genome sequence reveals the presence of an ORF (aq_455) that apparently encodes a SOR-like protein. A putative promoter (including −10 sequence ATAAAAA located 21 bases upstream from the methionine initiation codon and −35 sequence TTGAAA located 26 bases from the methionine codon) has been shown as the best fit to the E. coli σ70 consensus promoter sequence (McClure 1985). A distinct ribosome-binding sequence (AGGAGG) is not observed, except the purine-rich sequence GAGAG located seven bases upstream the ATG codon. No potential hairpin loop transcription termination signal is detected but the presence of a sequence rich in cytosine and poor in guanine and the identification of a rho-factor homologous gene (Washio et al. 1998) could suggest a rho-dependent termination. Alternatively, we can observe 20 bases downstream the stop codon, the pyrimidine-rich stretch (TTTTTATTT) already found in the A. ambivalens sor gene sequence (Kletzin 1992) that has been described as type I terminators in Sulfolobus acidocaldarius (Enori-Eta et al. 2000) and Methanococcus vannielii genes (Reiter et al. 1988; Wich et al. 1986). All these observations could suggest that the sor gene is not co-transcribed with other open reading frames.
Sequences comparison
The sequence identities (32%) between A. aeolicus and A. ambivalens SORs are shown in Fig. 1 (gray boxes). Despite an insertion in the 100 first residues of A. aeolicus sequence, the N-terminal half of SORs diverges less than the C-terminal half. Highly conserved regions between both SORs occur in the iron-binding site (with the conserved sequence motif H–X3–H–X23–E containing protein ligands) and in the residues lining the catalytic pocket such as the three cysteine residues (C36, C119 and C122) (Urich et al. 2005; Chen et al. 2005). The Phe residues (Phe 134 and Phe 142 in the C-terminal half of A. ambivalens) responsible for gating the chimney-entry pores (Urich et al. 2006) are respectively replaced by a Val and a Met which could increase the accessibility of the sulfur substrate into the catalytic center.

a Secondary structure prediction of SOR from A. aeolicus. Secondary structure of SORs from A. aeolicus and A. ambivalens are indicated by boxes for α helix and black boxes for β strand. Concensus secondary structure predictions of SOR from A. aeolicus was carried out using http://www.expasy.org (Psipred Protein Structure Prediction Server) and http://npsa-pbil.ibcp.fr/ (Predator) and secondary structure of SOR A. ambivalens was obtained from Urich et al. (2006, supplementary figures)
There are two structural domains in A. ambivalens formed by an internal β barrel (eight antiparallel β strands) partially surrounded by α helices (Urich et al. 2006). Secondary structure prediction of A. aeolicus SOR sequence revealed the substitution of β barrels six and eight observed in A. ambivalens by α helices in A. aeolicus (Fig. 1). This induces the loss, in A. aeolicus SOR, of the pseudosymmetry of the N and C terminal part described in A. ambivalens, which consists of two sets of four strands (Urich et al. 2006). However, the 30% identity between the second domain of A. aeolicus and A. ambivalens suggests the same protein ancestor. The substitution of β barrels by α helix in A. aeolicus probably generates a less stable protein which is in line with the presence of supplementary loop in A. aeolicus SOR sequence (Fig. 1).
Purification of wild-type SOR
To identify the enzyme which carries the SOR activity, different strategies of purification have been used on the soluble fraction. H2S production was detected in the membrane fraction as well as in the soluble fraction. All various protocols described in the literature and tested did not lead, in the case of A. aeolicus, to the pure enzyme. However, protocols based on exclusion chromatography and ion exchange or affinity columns allowed SOR enriched fractions to be obtained. The fractions with higher activity were combined to give the final preparation and SOR identification was made by N-terminal sequencing. Additional purification steps did not increase the purification factor of the enzyme but induced the loss of enzyme activity. Wild-type enriched SOR fraction was loaded on a BN gel and a band corresponding to 602 kDa was cut, hydrolysed by trypsine and peptides were analysed by MALDI-TOF mass spectrometry with a sequence coverage of 51% and 13 peptides for SOR identification. The identification of the SOR from the wild-type fraction and its partial purification shows for the first time the presence of the SOR in the homo-oligomer form in the bacterium, A. aeolicus.
The SOR fraction obtained has a specific activity of five units of reductase/mg of protein after three chromatographic steps (Table 1).
Cloning, heterologous production and purification of recombinant SOR from A. aeolicus
The sor gene, encoding the putative A. aeolicus SOR, was amplified by PCR and inserted into the pET22 expression vector. The resulting construct pETSOR, free of errors by DNA sequencing verification, was transformed in E. coli BL21-CodonPlus (DE3)-RIPL which contains extra-copies of tRNAs abundant in A. aeolicus and rare in E. coli. After induction, the protein was produced at high levels in the total crude extract. Although a significant fraction of the protein was found in inclusion bodies, analysis of the soluble fraction by SDS-PAGE and enzymatic assays revealed that soluble recombinant SOR was in sufficient amount for biochemical and biophysical studies; 28.8 mg of soluble protein was obtained from 10 l of culture after two steps of purification. A main band of 38 kDa, consistent with the calculated molecular mass deduced from the amino acid sequence of the SOR protein (37,674 kDa), was observed by SDS-polyacrylamide gel in the absence or presence of β-mercaptoethanol (Fig. 2). One other faint low-molecular-mass band (approximately 22 kDa) was observable but MALDI-TOF identification and N-terminal sequencing revealed that it is a proteolysis product of the 38 kDa SOR. The recombinant SOR obtained was pure, and no disulfide bonds between SOR subunits were observed. The same sample separated with non-denaturing BN gel (Fig. 3a, lane 1) showed a dominant band with an apparent molecular mass of 602 kDa corresponding to a homo-oligomer 16 subunits.

Separation of the purified recombinant A. aeolicus SOR on a SDS 10% polyacrylamide gel. Lane 1 20 μg of recombinant SOR. Lane 2 20 μg of recombinant SOR with β-mercaptoethanol. Lane 3 molecular mass markers (in kDa)

Oligomeric state of recombinant and wild-type SORs pre-incubated at 20 or 80°C. a Non-denaturing Blue Native gel stained with Coomassie blue of 30 μg of recombinant SOR pre-incubated before loading 30 min at 20°C (lane 1) or at 80°C (lane 2). b Immunoblotting experiments of recombinant and wild-type A. aeolicus SOR. Samples were incubated 30 min at 20°C (lanes 1 and 3) or at 80°C (lanes 2 and 4) before loading on a 10% non-denaturing native gel electrophoresis; 35 ng of recombinant SOR (lanes 1 and 2) and 10 μg A. aeolicus soluble extract (lanes 3 and 4) were loaded on the gel before detection by immunoblotting using anti-SOR antibodies
Active form of the SOR
Catalytic activity
The thermal properties of the recombinant enzyme were investigated by measuring the activity of the enzyme at different temperatures. The apparent optimal temperature of the SOR activity is 80°C and the enzyme has no significant activity at 20°C (the relative activity at 20°C is 3.6% of the maximal activity at 80°C; Fig. 5a, inset). The specific activity was measured from pH 3 to 9. The enzyme is active from pH 5.5 to 8 (data not shown).
The specific activities are 78.8 units of oxygenase/mg of protein and 3.05 units of reductase/mg of protein. No products were detected when the enzyme was incubated, in the absence of oxygen, under a hydrogen atmosphere. The specific activity obtained from reduction reaction was five times lower when the incubation was performed under an oxygen atmosphere (rather from air), due probably to the oxidation of H2S into thiosulfate (Kletzin 1989). SOR from A. aeolicus does not use thiosulfate or tetrathionate as substrate in our test conditions. As for SOR from A. ambivalens (Urich et al. 2004), the EPR spectrum of the recombinant enzyme showed a signal at g = 4.26 (data not shown) and is consistent with the presence of a mononuclear non-haem iron centre in the ferric high-spin state (Urich et al. 2006).
Oligomeric state of the SOR at various temperatures
Various bands were detected by BN gels (Fig. 3a, lane 1) suggesting the existence of various oligomeric forms of SOR. Gel filtrations chromatography experiments did not allowed getting more homogenous enzyme suggesting equilibrium between the different forms of the enzyme. To check if the oligomeric state of the enzyme is affected by temperature, non-denaturing blue-native gels were made with the recombinant enzyme pre-incubated at 20°C (Fig. 3a, lane 1) or with the enzyme pre-incubated at 80°C (Fig. 3a, lane 2). The prevalent band, whatever the pre-incubation temperature, is the one around 602 kDa. Several bands of lesser intensity are visible at 20°C (Fig. 3a, lane 1) and correspond to different oligomeric states of the enzyme (32, 26, 12, 2 or 1 subunits). Quantification of the different bands shows that the 16 mers represents the major form (77.2%). Both, the 12 and 26 mers are present in a same amount while the dimeric form represents 14% of the total protein. At 80°C, only two minor bands are observed, corresponding to the 32-mer and dimer forms (Fig. 3a, lane 2). Analysis of the recombinant and wild-type SORs by Western-blot experiments showed, whatever the sample, that incubation induce a more homogeneous form of the enzyme at 80°C than at 20°C (Fig. 3b). The 16-mer is the major form whatever the temperature and the enzyme used (Fig. 3b). To study the activity of the different oligomeric forms, we developed a new method for the detection of the SOR activity based on polysulfide consumption at 80°C by SOR enzyme embedded on native polyacrylamide gel. The consumption of polysulfide by the SOR enzyme is detected by the apparition of a white band. Whatever the sample, the band of 602 kDa is active (Fig. 4, lanes 3 and 4). At 80°C, only this active band is detected (Fig. 4, lane 4). An additional band at 480 kDa (12 subunits) exhibits an activity when enzyme is pre-incubated at 20°C and tested at 80°C (Fig. 4, lane 3). The relative low intensity of the activity detected compared to the intensity the band stained with coomassie blue, is explained by the low specific activity of SOR enzyme. If the entire enzyme is active, 3 μmol of H2S/min/mg are produced. When enzyme is pre-incubated at 20°C, the band at 602 kDa presents less activity (Fig. 4, lane 3) than the same band obtained with enzyme previously pre-incubated at 80°C (Fig. 4, lane 4). Other homo-oligomers of SOR of smaller sizes and mainly present after pre-incubation at 20°C (Fig. 4, lane 2) were inactive (Fig. 4, lane 3). Quantification of the corresponding bands on BN gel (see previously) shows that the inactivity of small oligomers is not due to a too low amount of proteins as 480 kDa band is less concentrated but exhibits an activity on the gel.

Detection of SOR activity on BN gel; 30 μg of recombinant SOR was pre-incubated 30 min at 20 or 80°C before loading on a BN gel. Lane 1 molecular marker. Lane 2 recombinant enzyme pre-incubated at 20°C and stained with Coomassie blue. Lanes 3 and 4 polysulfide was used as electron donor and 2,3,5-triphenyltetrazolium chloride as electron acceptor for detection of SOR activity pre-incubated at 20°C (lane 3) or at 80°C (lane 4). The activity was tested at 80°C
These results indicate (1) incubation at high temperature induces more homogenous form of the enzyme; (2) the acquisition of an “active conformation” induced by the temperature seems to be more important than the subunit number. They also indicate that the wild-type and recombinant SOR behave equally with respect to the temperature with the presence of a 16 homo-oligomer (band of 602 kDa prevalent whatever the temperature).
Spectroscopic studies of the inactive and active conformations
The Arrhenius plot (Fig. 5a) is biphasic, with a break point at 55°C, and plot-calculated activation energies were 3 and 14.7 kcal/mol which are characteristic of two distinct conformations with a different catalytic competence (Segel 1993).

a Thermal properties of recombinant SOR. Arrhenius plot was obtained by measuring the activity of recombinant SOR from 20 to 90°C (inset). Temperature dependence of activity. b CD spectroscopy in the far-UV. Far-UV spectra (from 240 to 200 nm) of the recombinant SOR measured at 293 K (dotted line) and 353 K (solid line). c CD spectroscopy in the near-UV. Ellipticity variation was recorded at 260 nm from 273 to 353°K (filled circle) and from 353 to 273°K (×) at 1°C/min. Each measure was done three times. Data analysis was done as described in Material and methods. Inset near-UV spectra (from 250 to 340 nm) at 293 K (black line) and 353 K (grey line)
To confirm this change of conformation by spectroscopic method, we performed CD experiments. CD spectra were recorded at 20 and 80°C in the far UV (Fig. 5b) and near UV (Fig. 5c, inset) regions. At 20°C, CD spectra of the recombinant protein in the far and near UV regions (Fig. 5b, c, inset) demonstrated that the protein is folded into a well-defined structure. Particularly, the presence of a signal different from zero in the near UV was a good indication that the form is folded at 20°C (Fig. 5c, inset) (Manning and Woody 1989). The spectrum performed at 20°C in the far UV (Fig. 5b) shows important modifications compared to the simulated theoretical spectrum for a protein (30% helix and 16% β) probably because of the presence of chromophores like aromatic “W101, Y109, F112, F116 F123” in the α helix 3 and the 3 proline clusters P142P143, P247P248, P315P316 that might perturb the far UV spectrum (Andrade et al. 1993). In contrast, secondary-structure analysis of the active SOR performed from the far UV spectrum obtained at 80°C (Fig. 5b), resulted in 37% of α-helix and 16% of β-sheet and 47% random which is in accordance with secondary-predictions values.
Spectra comparison (Fig. 5c, inset) in near UV between 20 and 80°C shows a significant positive absorption centred at 266 nm at 20°C which decreases with a shift at 277 nm at 80°C due to a lower influence of aromatic residues. This change suggests the existence of sensitive conformational modifications linked to the temperature probably in line with the acquisition of the active form of SOR. The ellipticity variation from 20 to 80°C is presented in Fig. 5c. Values of Tm were extracted from the melting curves by fitting the data to the equation reported by Tan et al. (1995). Tm values are 45°C ± 3.2 and 70°C ± 1.68. The first transition is in accordance with the break observed in Arrhenius plot. A total reversibility was observed when temperature was shifted again from 80 to 20°C suggesting a great flexibility of the enzyme and identical conformational steps from 20 to 80°C and from 80 to 20°C. Ellipticity measured at 222 nm showed no denaturation process from 20 to 80°C.
Altogether, these results show that the temperature increase to 80°C induces a change of conformation of SOR required for getting the active enzyme form. At 20°C the SOR is mainly present in the 16 subunits oligomeric form as at 80°C but in an inactive conformational state.
Discussion
SOR has been, to date, mainly detected in acidophilic archaea (Acidianus species, Sulfolobus tokodaii, Picrophilus torridus and Ferroplasma acidarmanus) and recently detected in one bacterial organism Acidithiobacillus sp. Strain SM-1 (Chen et al. 2007). Here, we report for the first time the characterisation and a biophysical study of a bacterial SOR, A. aeolicus. Analysis of genomes from other Aquificales such as Persephonella marina, Sulfurihydrogenibium yellowstonense and Sulfurihydrogenibium azorense, has not allowed, to date, to identify a gene encoding SOR.
SOR from A. aeolicus and Archaea are very similar in subunit size, the catalytic pocket composition and the oxidised reactions products (sulfite and thiosulfate) and reduced product (hydrogen sulfide). The oxygenase activity obtained from A. aeolicus SOR is approximately tenfold higher than SOR from Acidianus species and could be explained by amino-acid substitutions in the chimney-entry pore resulting in an increasing of the accessibility of the sulfur substrate into the catalytic center of A. aeolicus enzyme.
Determination of the size of the native and recombinant bacterial SORs by BN gel experiments revealed the presence of a predominantly 602 kDa homopolymer composed of 16 subunits. Even if BN gel experiment does not allow to determine the exact molecular weight of the enzyme, comparison of SORs subunit contents using this same technique shows that the subunit number of the A. aeolicus SOR (16) is smaller than the A. ambivalens SOR [20 by BN gel experiment (Urich et al. 2004) and 24 subunits by crystallographic determination (Urich et al. 2006)].
Except Ferroplasma acidarmanus in which sor is probably a pseudogene, all sor genes have been detected in thermophilic and hyperthermophilic organisms (Dopson et al. 2004). SORs from A. ambivalens (Urich et al. 2004) and A. tengchongensis (Sun et al. 2003) are inactive at mesophilic temperatures and significantly active from approximately 50–100°C. The A. aeolicus SOR is also inactive at 20°C. In order to understand the factors responsible for getting the activity at high temperature of this bacterial SOR, various experiments have been performed on the inactive (20°C) and active form (80°C). Different oligomeric forms of the enzymes are visible at 20 and 80°C but increasing the temperature induces more homogeneous form of the enzyme. Development of a new method for detection of the SOR activity on BN gels allows identifying the 16 mers as the active form and shows that the intact quaternary structure is indispensable for enzyme activity considering the absence of activity in the dissociated forms (subunit number of 1, 2, 4 or 10). Moreover, these results indicate that the inactivity of the SOR at 20°C does not result from a dissociation of the oligomer because the 16-mers is mainly present at 20°C as at 80°C. Spectroscopic data reveal a change of conformation linked to the temperature and indispensable for getting the active form. We can postulate that the lack of activity of the enzyme at 20°C derives either from an inability of substrate to reach the catalytic center or from an inadequate conformation of the catalytic pocket to allow catalysis.
Attempts to obtain a model of the 3D structure of A. aeolicus SOR from the A. ambivalens X-ray structure failed due to the insertion of two additional sequences in the bacterial enzyme: the first is composed of 13 residues and is located instead of the β-strand B3 of the A. ambivalens SOR and the second, smaller, is made of seven residues and is located in the C-terminal part. In A. ambivalens a ferredoxin fold with a repetition of (βαβ) topology has been described (Urich et al. 2006). The secondary structure prediction shows a substitution of β strand by α helix in the second structural domain of A. aeolicus SOR. This topology is loosed in the second domain of A. aeolicus SOR (C-terminal) and can lead to a more flexible structure. Without the 3D structure of the bacterial SOR, it is difficult to know the residues which could be involved in the structure stabilisation of the active form.
Our works on SOR from hyperthermophile bacteria show that the active form is induced by temperature even if the quaternary organisation is identical at each temperature. According to our spectroscopic results, we propose that increasing temperature induces conformational changes of the molecule allowing the enzyme to adopt an active conformation.
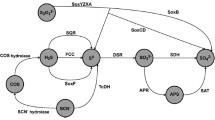
In A. ambivalens, it has been demonstrated that SOR interacts with the membranous thiosulfate:quinone oxidoreductase (TQO), elicited in A. ambivalens (Müller et al. 2004) responsible for the coupling between sulfur and oxygen metabolism. No gene encoding TQO has been detected in A. aeolicus genome. A possible model could be that the sulfide stem from the SOR would reduce the quinone pool via sulfide quinone reductase and the electrons from quinones would probably pass through the cytochrome bc 1 complex and cytochromes c555 to reduce molecular oxygen by a cytochrome c oxidase, as proposed by Nübel et al. (2000) and as it has been described in Acidothiobacillus (Wakai et al. 2004). A. aeolicus appears to be the sole organism, to our knowledge, containing in its genome genes encoding these two sulfur oxidation pathways: the bacterial SOX system and the SOR enzyme which is mainly found in the Archaeal domain. The exact function of the SOR is not yet understood but as it has been proposed previously (Urich et al. 2004), the presence of both sor and sox genes suggests that elemental sulfur is oxidized by the SOR, whereas the products of the reaction are oxidized by the SOX system or others enzymes. Thiosulfate and sulfite could be also oxidised, directly, by the periplasmic SOX system to form sulfate without the presence of free intermediary (Friedrich et al. 2005). Incomplete sox system without soxCD genes is detected in the genome from A. aeolicus (Deckert et al. 1998). The genes soxXYZAB, encode, in Parococcus pantotrophus, proteins that are required for sulfur-dependent cytochrome c reduction (Friedrich et al. 2001). Except for the SOX system none sulfite oxidase enzyme has been detected in the A. aeolicus genome.
It should be noted that the presence in the same organism of the SOR, SOX system and the SQR is completely unique and we are now investigating, by a physiological approach, the role of each in the sulfur metabolism.
Abbreviations
- A. aeolicus :
-
Aquifex aeolicus
- A. ambivalens :
-
Acidianus ambivalens
- S°:
-
Elemental sulfur
- SOR:
-
Sulfur oxygenase reductase
- SOX:
-
Sulfur oxidising enzyme system
- TOMES:
-
Thiosulfate oxidising multienzyme system
- TQOR:
-
Thiosulfate quinone oxidoreductase
- ORF:
-
Open reading frame
- CMC:
-
Carboxymethyl cellulose
- MALDI-TOF:
-
Matrix-assisted laser desorption ionization time-of-flight
- E. coli :
-
Escherichia coli
References
Andrade MA, Chacon P, Merelo JJ, Mora NF (1993) Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Prot eng 6:383–390
Brugna-Guiral M, Tron P, Nitschke W, Stetter KO, Burlat B, Guigliarelli B, Bruschi M, Giudici-Orticoni MT (2003) [NiFe] hydrogenases from the hyperthermophilic bacterium Aquifex aeolicus: properties, function, and phylogenetics. Extremophiles 7:145–157
Chen ZW, Jiang CY, She Q, Liu SJ, Zhou PJ (2005) Key role of cysteine residues in catalysis and subcellular localization of sulfur oxygenase-reductase of Acidianus tengchongensis. Appl Env Microbiol 71:621–628
Chen ZW, Liu YY, Wu JF, She Q, Jiang CY, Liu SJ (2007) Novel bacterial sulfur oxygenase reductases from bioreactors treating gold-bearing concentrates. Appl Microbiol Biotechnol 74:688–698
Deckert et al (1998) The complete genome of the hyperthermophilic bacterium Aquifex aeolicus. Nature 392:353–358
Dopson M, Barker-Austin C, Hind A, Bowman JP, Bond PL (2004) Characterization of Ferroplasma isolates and Ferroplasma acidarmanus sp. nov., extreme acidophiles from acid mine drainage and industrial bioleaching environments. Appl Environ Microbiol 70:2079–2088
Enori-Eta J, Gigot D, Thia-Toong TL, Glansdorff N, Charlier D (2000) Purification and characterization of Sa-lrp, a DNA-binding protein from the extreme thermoacidophilic archaeon Sulfolobus acidocaldarius homologous to the bacterial global transcriptional regulator Lrp. J bacteriol 182:3661–3672
Friedrich CG, Rother D, Bardischewsky F, Quentmeier A, Fischer J (2001) Oxidation of reduced inorganic sulfur compounds by bacteria: Emergence of a common mechanism? Appl Environ Microbiol 67:2873–2882
Friedrich CG, Bardischewsky F, Rother D, Quentmeier A, Fischer J (2005) Prokaryotic sulfur oxidation. Curr Opin Microbiol 8:253–259
Guermeur Y, Geourjon C, Gallinari P, Deleage G (1999) Improved performance in protein secondary structure prediction by inhomogeneous score combination. Bioinformatics 15:413–421
Guiral M, Tron P, Aubert C, Gloter A, Iobbi-Nivol C, Giudici-Orticoni M-T (2005) A membrane-bound multienzyme, hydrogen-oxidizing, and sulfur-reducing complex from the hyperthermophilic bacterium Aquifex aeolicus. J Biol Chem 280:42004–42015
Hedderich R, Klimmek O, Kröger A, Dirmeier R, Keller M, Stetter KO (1998) Anaerobic respiration with elemental sulfur and with disulfides. FEMS Microbiol Rev 22:353–381
Huber R, Stetter KO (2002) Aquificales. In: Encyclopedia of life sciences. John Wiley & Sons, Chichester, pp 1–6
Huber R, Wilharm T, Huber D, Trincone A, Burggraf S, König H, Rachel R, Rockinger I, Fricke H, Stetter KO (1992) Aquifex pyrophilus ge.nov.sp.nov. represents a novel group of marine hyperthermopilic hydrogen bacteria. Syst Appl Microbiol 15:340–351
Kletzin A (1989) Coupled enzymatic production of sulfite, thiosulfate, and hydrogen sulfide from sulfur: purification and properties of a sulfur oxygenase reductase from the facultatively anaerobic archaebacterium Desulfurolobus ambivalens. J Bacteriol 171:1638–1643
Kletzin A (1992) Molecular characterization of the sor gene, which encodes the sulfur oxygenase/reductase of the thermoacidophilic Archaeum Desulfurolobus ambivalens. J Bacteriol 174:5854–5859
Kletzin A, Urich T, Müller F, Bandeiras TM, Gomes CM (2004) Oxidation and reduction of elemental sulfur in themophilic bacteria. J Bioenerg Biomemb 36:77–91
Maniatis T, Fritsh E, Sambrook J (1982) Molecular cloning: a laboratory manual, Cold Spring Harbor Laboratory. Cold Spring Harbor
Manning MC, Woody RW (1989) Theoretical study of the contribution of aromatic side chains to the circular dichroism of basic bovine pancreatic trypsin inhibitor. Biochemistry 28:8609–8613
McClure WR (1985) Mechanism and control of transcription initiation in prokaryotes. Annu Rev Biochem 54:11–204
Morris MC, Mery J, Heitz A, Heitz F, Divita G (1999) Design and synthesis of a peptide derived from positions 195–244 of human cdc25C phosphatase. J Pept Sci 5:63–271
Müller FH, Bandeiras TM, Urich T, Texeira M, Gomes CM, Kletzin A (2004) Coupling of the pathway of sulfur oxydation to dioxygen reduction: characterization of a novel membrane-bound thiosulfate:quinone oxidoreductase. Mol Microbiol 53:1147–1160
Nübel T, Klughammer C, Huber R, Hauska G, Schütz M (2000) Sulfide:quinone oxidoreductase in membranes of the hyperthermophilic bacterium Aquifex aeolicus (VF5). Arch Microbiol 173:233–244
Reiter WD, Palm P, Zillig W (1988) Transcription termination in the archaebacterium Sulfolobus: signal structures and linkage to transcription initiation. Nucleic Acid Res 16:2445–2459
Rother D, Henrich HJ, Quentmeier A, Bardischewsky F, Friedrich CG (2001) Novel genes of the sox gene cluster, mutagenesis of the flavoprotein SoxF, and evidence for a general sulfur-oxidizing system in Paracoccus pantotrophus GB17. J Bacteriol 183:4499–4508
Schägger H, Von Jagow G (1991) Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem 199:223–231
Schägger H, Cramer A, Von Jagow G (1994) Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal Biochem 217:220–230
Segel IK (1993) Wiley Classics Library Edition, New York
Stetter KO, König H, Stackebrandt E (1983) Pyrodictium gen. nov., a new genus of submarine disc-shaped sulpur reducing archaebacteria growing optimally at 105°C. Syst Appl Microbiol 4:535–551
Sun CW, Chen ZW, He ZG, Zhou PJ, Liu SJ (2003) Purification and properties of the sulfur oxygenase/reductase from the acidothermophilic archaeon, Acidianus strain S5. Extremophiles 7:131–134
Suzuki I (1999) Oxidation of inorganic sulfur compounds: chemicals and enzymatic reactions. Can J Microbiol 45:97–105
Tan YJ, Oliveberg M, Davis B, Fersht R (1995) Perturbed pKA-values in the denatured states of proteins. J Biol Chem 254:980–992
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Urich T, Bandeiras TM, Leal SS, Albrecht T, Zimmermann P, Scholz C, Teixeira M, Gomes CM, Kletzin A (2004) The sulphur oxygenase reductase from Acidianus ambivalens is a multimeric protein containing a low-potential mononuclear non haem iron center. Biochem J 381:137–146
Urich T, Kroke A, Bauer C, Seyfarth K, Reuff M, Kletzin A (2005) Identification of core active site residues of the sulfur oxygenase reductase from Acidianus ambivalens by site-directed mutagenesis. FEMS Microbiol Lett 248:171–176
Urich T, Gomes CM, Kletzin A, Frazao C (2006) X-ray structure of a self-compartmentalizing sulfur cycle metalloenzyme. Science 311:996–1000
Wakai S, Kikumoto M, Kanao T, Kamimura K (2004) Involvement of sulfide:quinone oxidoreductase in sulfur oxidation of an acidophilic iron-oxidizing bacterium, Acidithiobacillus ferrooxidans NASF-1. Biosci Biotechnol Biochem 68:2519–2528
Washio T, Sasayama J, Tomita M (1998) Analysis of complete genomes suggests that many prokaryotes do not rely on hairpin formation in transcription termination. Nucleic Acid Res 26:5426–5463
Wich G, Hummel M, Jarsch M, Bar U, Böck A (1986) Transcription signals for stable RNA genes in Methanococcus. Nucleic Acid Res 14:2459–2479
Acknowledgments
We gratefully acknowledge the contribution of Marielle Bauzan (Institute of Microbiology and Structural Biology, CNRS, Marseilles, France) for growing bacteria; Bruno Gugliarelli for EPR experiments; and Danielle Moinier and Sabrina Lignon (Proteomic Analysis Center, Marseilles, France) for mass spectroscopy analysis. We also thank Alain Dolla, Wolfgang Nitschke, Mari Luz Cardenas and Athel Cornish-Bowden for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by K. Horikoshi.
Rights and permissions
About this article
Cite this article
Pelletier, N., Leroy, G., Guiral, M. et al. First characterisation of the active oligomer form of sulfur oxygenase reductase from the bacterium Aquifex aeolicus . Extremophiles 12, 205–215 (2008). https://doi.org/10.1007/s00792-007-0119-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-007-0119-5