
Abstract
Archaea utilize a branched modification of the classical Entner–Doudoroff (ED) pathway for sugar degradation. The semi-phosphorylative branch merges at the level of glyceraldehyde 3-phosphate (GAP) with the lower common shunt of the Emden-Meyerhof-Parnas pathway. In Sulfolobus solfataricus two different GAP converting enzymes—classical phosphorylating GAP dehydrogenase (GAPDH) and the non-phosphorylating GAPDH (GAPN)—were identified. In Sulfolobales the GAPN encoding gene is found adjacent to the ED gene cluster suggesting a function in the regulation of the semi-phosphorylative ED branch. The biochemical characterization of the recombinant GAPN of S. solfataricus revealed that—like the well-characterized GAPN from Thermoproteus tenax—the enzyme of S. solfataricus exhibits allosteric properties. However, both enzymes show some unexpected differences in co-substrate specificity as well as regulatory fine-tuning, which seem to reflect an adaptation to the different lifestyles of both organisms. Phylogenetic analyses and database searches in Archaea indicated a preferred distribution of GAPN (and/or GAP oxidoreductase) in hyperthermophilic Archaea supporting the previously suggested role of GAPN in metabolic thermoadaptation. This work suggests an important role of GAPN in the regulation of carbon degradation via modifications of the EMP and the branched ED pathway in hyperthermophilic Archaea.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Comparative studies of the carbohydrate metabolism in Archaea indicate that sugars are generally metabolized by modifications of the classical Entner–Doudoroff (ED) and Embden-Meyerhof-Parnas (EMP) pathways that are operative in Bacteria and Eukarya (Ronimus and Morgan 2002; Siebers et al. 2004; Siebers and Schönheit 2005; Verhees et al. 2003). The archaeal pathways are characterized by the presence of numerous novel enzymes and enzyme families. Biochemical studies demonstrate that modifications of the EMP pathways are used in most anaerobic, fermentative Archaea (e.g. Pyrococcus furiosus), whereas ED modifications were identified in aerobic and facultatively anaerobic, heterotrophic Archaea (e.g. Sulfolobus solfataricus, Thermoplasma acidophilum, respectively). To date, the only Archaeon known to utilize both the EMP and the ED modification in parallel is the hyperthermophilic anaerobe Thermoproteus tenax (for a recent review, see Siebers and Schönheit 2005).
Initial biochemical studies in Archaea revealed the presence of the non-phosphorylative (np) ED pathway in (hyper)thermophiles (De Rosa et al. 1984; Siebers and Hensel 1993; Siebers et al. 1997; Selig et al. 1997) and the semi-phosphorylative (sp) ED pathway in halophilic archaea (Johnsen et al. 2001; Tomlinson et al. 1974). However, recent combinations of genomics-based and biochemical analyses demonstrated the presence of the branched ED pathway in all Archaea that utilize the ED pathway (Siebers et al. 2004; Ahmed et al. 2005; Jung and Lee 2005; Kehrer et al. 2007). The only exception known so far is Halobacterium sp. NRC-1, which harbors no known glycerate kinase homolog and thus seems to rely on the spED pathway for glucose degradation (Kehrer et al. 2007).
The thermo-acidophilic crenarchaeote Sulfolobus solfataricus is able to grow on a wide variety of sugars (e.g. starch, glucose, arabinose, fructose; Grogan 1989) and relies on the modified branched ED pathway for glucose catabolism (Ahmed et al. 2005). In the common shunt of the branched ED pathway, glucose dehydrogenase first oxidizes glucose to glucono-lactone (Giardina et al. 1986; Lamble et al. 2003), which is subsequently converted to gluconate either via a spontaneous reaction or via an enzymatic conversion by glucono-lactonase. Gluconate dehydratase then catalyzes the dehydration of gluconate to 2-keto-3-deoxygluconate (KDG) (Ahmed et al. 2005; Kim and Lee 2005; Lamble et al. 2004; Verhees et al. 2003). In the npED branch, the bifunctional 2-keto-3-deoxy-(6-phospho)-gluconate (KD(P)G) aldolase, which is a key player in both branches, cleaves KDG into pyruvate and glyceraldehyde (Buchanan et al. 1999; Lamble et al. 2003; Lamble et al. 2005; Ahmed et al. 2005). Glyceraldehyde is then oxidized to glycerate by glyceraldehyde dehydrogenase (Reher and Schönheit 2006; Jung and Lee 2006) or glyceraldehyde oxidoreductase (Kardinahl et al. 1999; Mukund and Adams 1991; Schicho et al. 1993; Selig and Schönheit 1994). The subsequent phosphorylation by glycerate kinase (MOFRL family) results in the formation of 2-phosphoglycerate (Kehrer et al. 2007; Reher et al. 2006). Finally, pyruvate is formed by the combined action of enolase and pyruvate kinase (De Rosa et al. 1984; Selig et al. 1997).
In the spED branch, phosphorylation occurs at the level of KDG by the action of two different types of KDG kinase. In T. tenax, Sulfolobales and Halobacteriales KDG kinases of the ribokinase-like superfamily [pfkB family carbohydrate kinase; PF00294, T. tenax (Ahmed et al. 2005), S. solfataricus (Ahmed et al. 2005, Lamble et al. 2005)] and in Thermoplasmatales KDG kinases of the the BadF/BadG/BcrA/BcrD ATPase family [PF01869; T. acidophilum (Jung and Lee 2005)] were identified and characterized. The resulting product 2-keto-3-deoxy-6-phoshogluconate (KDPG) is subsequently cleaved into pyruvate and glyceraldehyde-3-phosphate (GAP) by the bifunctional KD(P)G aldolase that is also active in the npED pathway and displays dual activity towards both phosphorylated (KDPG) and non-phosphorylated substrates (KDG). GAP is further processed via the common lower shunt of the EMP pathway finally forming a second molecule of pyruvate. Interestingly, this branched ED pathway was shown to be promiscuous for glucose and galactose in S. solfataricus (Lamble et al. 2003, 2004, 2005; Theodossis et al. 2004, Milburn et al. 2006).
For the oxidation of GAP, three different enzymes were reported in Archaea. There is the classical GAP dehydrogenase (GAPDH, phosphorylating, EC 1.2.1.13) and phosphoglycerate kinase couple, and there are two distinct types of non-phosphorylating enzymes: GAP dehydrogenase (GAPN, EC 1.2.1.9) and GAP oxidoreductase (GAPOR, EC 1.2.7.6). The latter two enzymes catalyze the unidirectional direct oxidation of GAP forming 3-phosphoglycerate but differ in their co-substrate specificity [pyridine nucleotides (GAPN), ferredoxin (GAPOR)] (Brunner et al. 1998; Mukund and Adams 1995; Siebers and Schönheit 2005; Van der Oost et al. 1998). Biochemical studies and transcriptional data in T. tenax and Pyrococcus furiosus, which harbor all three GAP converting enzymes, revealed that GAPN and GAPOR substitute for the anabolic enzyme couple NADP+-dependent GAPDH and phosphoglycerate kinase (PGK) in these hyperthermophiles at the expense of substrate-level phosphorylation (Brunner et al. 1998, 2001; Lorentzen et al. 2004; Schäfer and Schönheit 1993; Schut et al. 2003; Van der Oost et al. 1998). In S. solfataricus only two GAP converting enzymes, classical GAPDH and GAPN, were identified (Verhees et al. 2003).
Currently, the GAPN of T. tenax is the only characterized archaeal enzyme from this enzyme family. In T. tenax GAPN was identified as an important constituent of the modified EMP pathway, and a key function in the regulation of the pathway was reported. The enzyme was shown to catalyze the NAD(P)+-dependent unidirectional formation of 3-phosphoglycerate, and a sophisticated allosteric regulation by a number of metabolites was demonstrated (Brunner et al. 1998; Brunner and Hensel 2001; Lorentzen et al. 2004). The crystal structure of T. tenax GAPN in complex with both co-substrates, GAP and activating molecules was established (Lorentzen et al. 2004; Pohl et al. 2002). Phylogenetic analyses revealed that the GAPN family constitutes a distinct subfamily within the diverse aldehyde dehydrogenase (ALDH) superfamily (PF00171) members of which have been described in all three domains of life (Brunner et al. 1998). ALDHs catalyze the NAD(P)+-dependent oxidation of aldehydes to the corresponding carboxylic acids and play an important role in several cellular processes [e.g. glycolysis, detoxification, embryogenic development (Yoshida et al. 1998)]. More recently, new archaeal members of the ALDH superfamily were characterized: (1) glyceraldehyde dehydrogenase [Picrophilus torridus (Reher and Schönheit 2006], T. acidophilum (Jung and Lee 2006; Reher and Schönheit 2006), a constituent of the npED branch, and (2) 2,5-dioxopentanoate dehydrogenase involved in the catabolic d-arabinose pathway (Brouns et al. 2006).
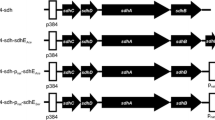
Strikingly, genes of the branched ED pathway (gluconate dehydratase, KDG kinase and KD(P)G aldolase) were found to be clustered in the genome of S. solfataricus and T. tenax. Parts of this gene cluster were also found in S. tokodaii, S. acidocaldarius and Halobacterium sp. NRC-1 (Ahmed et al. 2005). In all three Sulfolobus species, a gene encoding non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase (GAPN) was detected adjacent to this gene cluster (Fig. 1), suggesting a prominent role of this enzyme in the spED branch. The predicted GAPN activity was confirmed for the recombinant S. solfataricus enzyme in E. coli cell extracts after heat precipitation (Ahmed et al. 2005).

Genome context analysis of identified ED gene clusters in Archaea. Partially conserved ED gene cluster encoded by multiple archaeal genomes, comprising genes encoding gluconate dehydratase (gad), KD(P)G aldolase (kdgA) and KDG kinase (kdgK). In the genomes of three Sulfolobus species sequences, the gapN genes are located directly downstream of this gene cluster, suggesting a function in the spED branch in these organisms
Here, we present the molecular and biochemical characterization of the GAPN of S. solfataricus (Sso-GAPN) and provide evidence that GAPN plays an important role in the regulation of the spED branch in this organism. Furthermore, we discuss some phylogenetic aspects, the distribution of the three GAP converting enzymes (GAPDH, GAPN, GAPOR) in Archaea and present physiological implications of our results for archaeal glycolysis.
Materials and methods
Strains and growth conditions
Cultures of S. solfataricus P2 (DSM 1617, Zillig et al. 1980) were grown as reported previously (Brinkman et al. 2002). The different carbon sources were added to a final concentration of 0.2% (w/v). Escherichia coli strains DH5α (Life Technologies), BL21(DE3) CodonPlus (Novagen) for cloning and expression studies were grown under standard conditions (Sambrook et al. 1989) following the instructions of the manufacturer.
(Bio)chemicals and enzymes
If not indicated otherwise, (bio)chemicals and enzymes were purchased from Sigma-Aldrich, VWR International or Roche Diagnostics GmbH in analytical grade. (d, l)-Glyceraldehyde 3-phosphate was purchased from Sigma.
Heterologous expression and protein purification
Heterologous expression of the S. solfataricus GAPN was performed as reported previously (Ahmed et al. 2005), with the exception that BL21(DE3) CodonPlus was used as expression host. For protein purification, cells of an E. coli expression culture (7.5 g wet weight) were resuspended in 15 ml resuspension buffer [100 mM HEPES/KOH (pH 7.5) containing 7.5 mM dithiothreitol], followed by three passages through a French pressure cell at 150 MPa. Cell debris and unbroken cells were removed by centrifugation (60,000 × g for 30 min at 4°C). The crude cell extract was diluted 1:1 with resuspension buffer and initial purification of Sso-GAPN was performed by heat incubation of the diluted crude cell extract (30 min at 85°C). Subsequently, precipitated proteins were removed by centrifugation (60,000 × g for 30 min at 4°C), and the cleared lysate was dialyzed overnight against 50 mM HEPES/KOH (pH 7.0, 70°C), 7.5 mM dithiothreitol, 300 mM KCl (2-liter volume, 4°C). The dialyzed lysate was concentrated and applied to a HiLoad 26/60 Superdex 200 prep grade column (Amersham Biosciences) which was equilibrated with dialysis buffer. Thermostable GAPN activity eluted as one single peak and SDS-PAGE analysis revealed a single band with a relative molecular mass of approximately 57 kDa, which is in good agreement with the calculated mass of a single subunit (56.927 Da). Fractions containing the homogeneous enzyme were pooled and used for subsequent protein characterization. The native molecular mass was determined by gel filtration on a HiLoad 26/60 Superdex 200 prep grade column (Amersham Biosciences) using the same running conditions as for enzyme purification. Five different runs in absence and presence of standard proteins were performed [Standards: ferritin type I (horse spleen, 443 kDa), alcohol dehydrogenase (yeast, 148 kDa), d-lactate dehydrogenase (Lactobacillus leichmanii, 78 kDa), and cytochrome c (bovine heart, 12.5 kDa)]. Protein concentration was determined using the reagent kit from BioRad (München, Germany) and bovine serum albumin as standard. 7.5 g of wet cells of recombinant E. coli yielded 8.4 mg of homogenous GAPN with a specific activity of 3.74 U mg−1.
Enzyme assays and determination of kinetic parameters
GAPN activity was determined using a continuous enzyme assay at 70°C. The GAPN standard enzyme assay was performed in assay buffer [90 mM HEPES/KOH (pH 7.0, 70°C), 160 mM KCl and 2 mM NADP+ or NAD+] in a final volume of 1 ml. Reactions were started upon addition of d,l-GAP (final concentration 5 mM) in the presence of purified Sso-GAPN (6 μg ml−1). Enzymatic activities were measured by monitoring the increase of NADPH or NADH at 340 nm (ɛNADPH at 70°C = 5.71 mM−1 cm−1, ɛNADH at 70°C = 5.8 mM−1 cm−1). For each assay three independent measurements were performed and the experimental error was determined. Calculation of the kinetic parameters (V max and K m) were performed by iterative curve-fitting (Hanes) using the program Origin (Microcal Software Inc.). Effector studies were performed in the presence of half-saturating concentrations of NADP+ or NAD+ (0.1 or 20 mM, respectively) and d,l-GAP (500 μM) with 18 μg GAPN. The following metabolites in the concentration range of (0.001–1 mM) were tested: Glucose 1-phosphate, fructose 6-phosphate, glucose, gluconate, galactonate, KDG, glyceraldehyde, glycerate, pyruvate, AMP, ADP and ATP. The effect of glucose 1-phosphate on kinetic parameters was studied in the presence of 0.01 mM effector.
Transcript analysis
Total RNA was isolated from S. solfataricus P2 mid-log cultures (A600 = 0.5) grown on d-glucose, d-arbinose and tryptone using an RNeasy kit (QIAgen). 50 ml of culture was washed in 1 ml of medium and resuspended in 100 μl of TE (10 mM Tris–HCl, 1 mM EDTA, pH 8.0). After addition of 5 μl of 10% Triton X-100, the RNA was further purified according to the manufacturer’s instructions, except that genomic DNA was sheared through a 0.45-mm needle before the sample was applied onto a spin column. Columns were eluted twice with 50 μl of water. The transcription start sites were mapped for the transcripts of gapN and the gad-kdgA-kdgK operon of S. solfataricus, respectively. Primer extension analysis was performed using the following radiolabeled antisense oligonucleotides: 5′-CCATTTTCCGTAATGACCCTTGTGAC-3′, for the S. solfataricus gad gene and 5′-CTGATCCACTGACCCGATAGATAGG-3′ for the S. solfataricus gapN gene. Primer extension reactions were performed using the AMV-RT Kit (Promega) according to the manufacturer’s instructions. For the primer extension reaction, 30 μg of total RNA and 2.5 ng of radiolabeled oligonucleotide were resuspended in 2× AMV-RT buffer (Promega) in a final volume of 25 μl. Samples were heated to 70°C for 10 min and slowly cooled to room temperature. MgCl2, dNTPs, RNasin, and AMV-RT (Promega) were added to a concentration of 5 mM, 0.4 mM, 0.8 U μl−1, and 0.4 U μl−1, respectively, in a final volume of 50 μl. The samples were incubated at 42°C for 30 min, extracted with phenol/chloroform, precipitated with ethanol and resuspended in formamide loading buffer. The primer extension product was analyzed on an 8% denaturing sequencing gel along with a sequence ladder that was generated using the same radiolabeled oligonucleotides.
Phylogenetic analyses
A multiple alignment of GAPN-like protein sequences was constructed using MUSCLE (Edgar 2004) followed by manual adjustments based on PSI-BLAST results. Protein secondary structure was assigned according to the resolved structure of T. tenax GAPN (Lorentzen et al. 2004). The alignment was used for the reconstruction of a phylogenetic tree by calculating the evolutionary distances using the JTT model of amino acid evolution (Jones et al. 1992) as implemented in the PHYML package (Guindon and Gascuel 2003). Bootstrap analysis was performed for the maximum likelihood tree as implemented in PHYML (Guindon and Gascuel 2003).
Results and discussion
The presence of the branched ED pathway in Archaea rises questions about its regulation and more profound about its physiological function. Although several enzymes of this pathway have been characterized, their regulatory properties have not been addressed. So far the glycerate kinase of T. tenax has been the only enzyme that was shown to exhibit regulatory properties (competitive inhibition via ADP) and regulation by the energy charge of the cell has been suggested (Kehrer et al. 2007). During the re-evaluation of the modified ED pathway in Archaea, we encountered a partially conserved gene cluster composed of genes encoding gluconate dehydratase (gad) (Lamble et al. 2004; Kim and Lee 2005; Ahmed et al. 2005), KDG kinase (kdgK) (Ahmed et al. 2005; Lamble et al. 2005) and KD(P)G aldolase (kdgA) (Ahmed et al. 2005; Buchanan et al. 1999; Lamble et al. 2005; Theodossis et al. 2004) (Fig. 1). Interestingly, in the genomes of all three Sulfolobus species, for which sequence information is available (S. solfataricus, S. tokodaii and S. acidocaldarius), a gene encoding a homolog of T. tenax GAPN (Ttx-GAPN) (Ahmed et al. 2005) was identified directly downstream of this ED gene cluster. This conserved clustering suggested an important function of GAPN in the regulation of the spED branch in these organisms. The encoding gene of S. solfataricus (SSO3194) was cloned, the recombinant gene product was enriched by heat precipitation and the predicted GAPN activity was confirmed previously (Ahmed et al. 2005). In order to address the suggested role of GAPN in the regulation of the spED branch of S. solfataricus, the enzyme was purified and analyzed for its enzymatic and regulatory properties.
Genome organization and transcription start sites in S. solfataricus
In S. solfataricus the gad, kdgA, kdgK, and gapN genes all reside in the same coding strand (Fig. 2a), being separated by 2, 9, and 39 bp, respectively. Putative promoter elements were only identified in the upstream regions of the gad and gapN gene suggesting a polycistronic transcript of the gad–kdgA–kdgK genes and a monocistronic transcript of the gapN gene (Fig. 2c). For a more accurate assignment of the promoter region in S. solfataricus, the transcription starts of the gad–kdgA–kdgK and gapN mRNA of S. solfataricus, respectively, were determined by primer extension analyses (Fig. 2b). As shown in Fig. 2b, c transcription of both transcripts is initiated at the thymidine (T) immediately in front of start codon ATG, thus lacking Shine-Dalgarno sequences. Separate transcription start sites for the internal kdgA and kdgK genes could not be detected (data not shown). The assignment of crenarchaeal consensus promoter sequences in front of the first gene of an operon (gad) and in front of downstream located single genes (gapN), the absence of Shine-Dalgarno sequences upstream of the first gene and subsequent translation via leaderless transcripts is in good agreement with previous studies in S. solfataricus (Condo et al. 1999; Tolstrup et al. 2000) and other Crenarchaea [T. tenax (Schramm et al. 2000; Siebers et al. 2001, 2004), Pyrobaculum aerophilum (Slupska et al. 2001)].

Transcriptional analysis of the gapN gene and the ED gene cluster in S. solfataricus. a Genomic organization in S. solfataricus. Open reading frames and their orientation are indicated by arrows. The transcription start sites are indicated by arrows. b Determination of transcription start sites of the gapN gene (left panel) and ED operon (right panel; gad gene, first gene of the ED operon). Mapping of the start sites revealed that transcription was initiated at the −1 position for both transcripts (relatively to the translational start site), giving rise to leaderless transcripts. ‘P’ denotes lanes containing the primer extension products. c Analysis of the upstream regions of the gapN and gad revealed the presence of putative basal transcription factor binding sites. Putative TATA-box (boxed) and BRE sites (underlined) are indicated. Transcription start sites are indicated with an arrow, and the translational start site (ATG) is indicated in bold
The transcription start points were confirmed by analysis of RNA samples derived from cultures grown on different carbon sources (S. solfataricus cells grown on d-glucose, d-arabinose and tryptone; data not shown). These first studies revealed only minor differences in transcript abundance, suggesting a constitutive expression of the ED gene cluster and the gapN gene under the chosen growth conditions. These results were confirmed by the recently combined DNA microarray and proteomics analyses, which revealed a slight up-regulation of GAPN on glucose (compared to tryptone/yeast extract) and constitutive production of the operon-encoded enzymes (Snijders et al. 2006). In summary, primer extension analysis of RNA isolated from S. solfataricus strongly suggests that the gapN gene is an independent transcriptional unit, transcribed separately from the gad–kdgA–kdgK gene cluster (Fig. 2a). The advantage of two separate promoters might be an increased metabolic flexibility. Previously, it has been suggested that in S. solfataricus the catabolic EMP pathway is utilized for fructose degradation or other catabolic pathways that proceed via fructose or fructose 6-phosphate (She et al. 2001). Therefore an independent regulation of specific genes of the branched ED pathway as well as genes of the common lower shunt of the EMP pathway seem to be favorable in order to adapt and respond to different carbon sources.
Enzyme characterization
The GAPN gene was cloned and expressed in BL21 (DE3) CodonPlus using the pET expression vector system. The recombinant enzyme was purified from E. coli crude extracts by heat precipitation and gel filtration. GAPN catalyzes the irreversible, non-phosphorylating oxidation of GAP to 3-phosphoglycerate. The S. solfataricus GAPN activity was determined in a continuous assay at 70°C monitoring the formation of NADPH or NADH at 340 nm. The Sso-GAPN follows classical Michaelis-Menten kinetics for NADP+ (K m of 0.09 ± 0.01 mM and V max of 4.61 ± 0.09 U mg−1 protein) and d,l-GAP (K m of 0.51 ± 0.04 mM and a V max of 4.62 ± 0.09 U mg−1 protein) (Fig. 3). However, in contrast to the NADP+-dependent reaction the enzyme shows only negligible activity with NAD+ (Table 1). The NAD+-dependent reaction of Sso-GAPN shows no saturation at NAD+ concentrations up to 50 mM. The highest enzyme activity was observed at 50 mM NAD+ (0.61 U mg−1 protein), which is 7- to 8-fold lower than the V max observed using NADP+ as co-factor. In addition, the apparent K m value for NAD+ is at least 200-fold higher than for NADP+ (Table 1). Furthermore, as reported previously for the NADP+-dependent reaction of the Ttx-GAPN, the saturation kinetics of the NAD+-dependent reaction of Sso-GAPN shows a bumpy curve with several pronounced intermediate plateaus (Lorentzen et al. 2004) (data not shown). Similar non-linear saturation kinetics have been described for several enzymes with more than two ligand-binding sites (Corwin and Fanning 1968; Gotz and Schleifer 1975; LeJohn and Jackson 1968). However, the molecular basis underlying this behavior is still unclear. Therefore, NADP+ seems to represent the physiological co-substrate for the Sso-GAPN. In this respect, Sso-GAPN resembles most GAPNs characterized to date, which also prefer NADP+ more than NAD+ as co-factor (Habenicht 1997; Perozich et al. 2000). Strikingly, this apparent co-factor preference of Sso-GAPN is different for its close homolog in T. tenax (56% identity). The Ttx-GAPN exhibits a 2.6-fold higher velocity and 6.5-fold higher affinity using NAD+ as co-factor compared to NADP+ in absence of activators (see below) (Lorentzen et al. 2004) (Table 1). Furthermore, NADP+ was shown to be a competitive inhibitor of the NAD+-dependent reaction of Ttx-GAPN (apparent K D of 1.0 μM) (Brunner et al. 1998).

Kinetic properties of the GAPN of S. solfataricus. The GAPN activity was determined in a continuous assay at 70°C. The dependence of the specific enzyme activity on GAP [in absence (a) and presence of glucose 1-phosphate (G1P) (b)] and NADP+ [in absence (c) and presence of G1P (d)] concentration is shown. The enzyme follows classical Michaelis-Menten kinetics for both co-substrate and substrate in the absence and presence of the activator G1P (0.01 mM). The insert shows the linear transformation according to Hanes
In order to unravel the role of GAPN in the regulation of the branched ED pathway, effector studies were performed in the presence of half-saturating concentrations of GAP and NADP+. In addition to effectors (metabolites) reported to influence the Ttx-GAPN (see Material and methods; Brunner et al. 1998; Lorentzen et al. 2004) also intermediates of the branched ED pathway were analyzed (concentration 0.001–1 mM). As reported for the T. tenax enzyme, the most efficient effector identified is glucose 1-phosphate (G1P) (14-fold activation at 0.1 mM G1P). Of the other metabolites tested, minor stimulatory effects (1.1- to 1.2-fold) were observed in the presence of fructose 6-phosphate, AMP and pyruvate. ATP, gluconate and galactonate were found to slightly (1.2- to1.3-fold) inhibit Sso-GAPN activity, whereas glyceraldehyde and ADP have no effect on GAPN activity (data not shown). These observations are contrasting with the strong allosteric properties of Ttx-GAPN, which is activated 2.2-, 2.8- and 2.5-fold upon addition of F6P, AMP or ADP, respectively, when NADP+ is used as co-factor (Lorentzen et al. 2004).
Due to these obvious differences with respect to co-substrate binding and regulatory properties of both archaeal GAPNs, the effect of G1P on Sso-GAPN was analyzed in more detail. The velocity of Sso-GAPN in the presence of NADP+ increased by about 4.1-fold on addition of G1P (0.01 mM) and a significant activation of V max (2-fold to 3-fold) by G1P was observed in the presence of 50 mM NAD+ as co-factor (Table 1, Fig. 3). Strikingly, the affinity of Sso-GAPN for both co-factors NAD+ and NADP+ as well as for the substrate GAP was not affected upon addition of G1P. This finding represents an important difference to the T. tenax enzyme, which shows a similar activation of velocity (3-fold) for the NADP+-dependent reaction, but in addition a dramatic effect on co-substrate affinity. In presence of G1P (0.1 mM), a 200-fold increase in affinity for NADP+ is observed. In contrast, the NAD+-dependent reaction of the T. tenax enzyme revealed no difference in velocity, and the affinity for NAD+ increased by about 8-fold in presence of G1P (Lorentzen et al. 2004) (Table 1). Due to this dramatic change of affinity for NADP+ in presence of G1P, the authors suggest that NADP+ is the preferred co-substrate in the presence of activators for the Ttx-GAPN. In the absence of activators, NADP+ acts as an effective competitive inhibitor of the NAD+-dependent reaction (Brunner et al. 1998; Lorentzen et al. 2004). The effect of G1P on GAPN affinity for GAP was not analyzed for the T. tenax enzyme.
Close inspection of the 3D structure that has been solved for Ttx-GAPN revealed obvious differences between the Ttx- and Sso-GAPNs regarding some residues that constitute the NAD(P)+-binding pocket, which might account for the apparent difference in co-factor preference. The side chains of three residues (T165, S193 and I194) involved in co-substrate binding are replaced by valine, proline and serine residues in Sso-GAPN (V172, P200 and S201, respectively, Fig. 4; Lorentzen et al. 2004). S193 and I194 have been shown to tightly bind the 2′-phosphate group of NADPH (Lorentzen et al. 2004). No obvious differences are observed in the remaining residues that make up the NADH/NADPH binding pocket, as well as the activator binding residues and the active site (Fig. 4). How the observed structural differences in the co-factor binding pocket of GAPN correlate with the detected differences in co-factor affinity remains to be established.

Multiple sequence alignment of characterized archaeal members of the aldehyde dehydrogenase superfamily (GAPN, S. solfataricus and T. tenax; glyceraldhyde dehydrogenase, (GADH) T. acidophilum and P. torridus; and 2,5-dioxypentanoate dehydrogenase, (DopDH) S. solfataricus) and GAPN-paralogs identified in the genome of S. solfataricus. As a reference, the well-characterized S. mutans GAPN is also included in the alignment. Residues that are conserved >90% across the sequences that are included in the alignment are shaded in gray. Secondary structure was assigned according to the resolved structure of Ttx-GAPN (PDB: 1UXN), residues part of an α-helix and a β-strand are denoted with ‘E’ and ‘S’, respectively. Above the alignment, residues that form the active site are indicated with an asterisk (*), whereas residues that form binding pockets of the activators and co-factor are indicated by ‘#’ and ‘¥’, respectively (Lorentzen et al. 2004). The 90% consensus sequence of the aligned protein sequences is depicted below the alignment, using the following amino acid classes: - negative residues (DE); + positive residues (KRH); a aromatic residues (FYWH); t tiny residues (ACGS); p polar residues (DEHKRNQCSTYW) and h hydrophilic residues (ILVMFYWHKTCGAP), of which the aliphatic residues (ILV) are indicated with l. For species abbreviations, see Fig. 5
In summary, the enzymatic studies reveal that, like the T. tenax GAPN, the enzyme of S. solfataricus represents a non-phosphorylating, allosteric GAPDH that catalyzes the irreversible oxidation of glyceraldehyde 3-phosphate to 3-phosphoglycerate. However, both enzymes differ in co-substrate specificity and regulatory properties. Although Ttx-GAPN is able to use both NAD+ and—in the presence of activators even more efficiently—NADP+ (Lorentzen et al. 2004) as co-substrate, Sso-GAPN shows only redundant activity with NAD+ as co-substrate and thus NADP+ seems to be the physiological co-substrate. Furthermore, the allosteric potential of Sso-GAPN is reduced compared to the T. tenax enzyme and, as demonstrated for G1P, the influence of activators on the enzyme differs significantly with respect to co-substrate binding. These studies indicate a different fine-tuning of regulation for the T. tenax and the S. solfataricus GAPN, which might be explained by differences in the lifestyle of both organisms. The facultative heterotroph T. tenax seems to need a sophisticated regulation at the level of GAP in order to allow for effective gluconeogenesis under autotrophic growth conditions. T. tenax uses the anabolic EMP pathway for gluconeogenesis and the catabolic EMP pathway as well as the branched ED pathway for glycolysis. Under autotrophic growth conditions, the co-substrates of anabolic GAPDH (NADPH/NADP+) inhibit the catabolic, NAD+-dependent reaction of GAPN and permit gluconeogenesis. Only in the presence of activators, which signalize available carbon sources or low energy charge of the cell, NADP+ is the preferred co-substrate of GAPN, favouring glycolysis. This elaborate regulation avoids futile cycling at the level of GAP. In the heterotroph S. solfataricus, the situation is much more straightforward. S. solfataricus relies on the branched ED pathway for glucose degradation. The EMP pathway is supposed to be active only in gluconeogenetic direction allowing for glycogen formation and for the degradation of fructose or alternative substrates (e.g. sucrose; She et al. 2001). For the conversion of glucose to fructose 1,6-bisphosphate respective sugar kinases are missing. Like in T. tenax the activation of GAPN by G1P as intermediate of glycogen metabolism seems to be an important signal for enhanced carbon degradation. However, because of the absence of autotrophic growth, there seems to be no need for complex regulation of GAPN in this organism. According to this hypothesis it is tempting to speculate that the GAPN of the fermentative hyperthermophile Pyrococcus furiosus also exhibits reduced allosteric properties. However, this assumption still needs experimental confirmation.
Phylogenetic analyses
Database searches revealed GAPN homologs within several archaeal genomes with a preferred abundance in hyperthermophiles [e.g. Sulfolobales, Aeropyrum pernix, T. tenax, Thermococcus kodakaraensis, Pyrococcus furiosus, Halobacterium sp NRC-1, Fig. 5; Table 2]. In addition, four distant paralogs (SSO1218, SSO1629, SSO1842, SSO3117; Fig. 5) were identified in S. solfataricus. Phylogenetic analyses indicate that the archaeal GAPN homologs are part of the same orthologous cluster that includes the characterized bacterial GAPN from Streptococcus mutans (Boyd et al. 1995; Cobessi et al. 1999). All other GAPN homologs that are encoded by the S. solfataricus genome (SSO1629, SSO1842 SSO3117 and SSO1218) group outside of this cluster (Fig. 5). The only characterized paralog in S. solfataricus is SSO3117 for which 2,5-dioxopentanoate dehydrogenase (EC 1.2.1.26) activity was demonstrated (Brouns et al. 2006). The enzyme is a constituent of the catabolic d-arabinose pathway (pentose oxidation) in S. solfataricus, and significant activities with 2,5-dioxopentanoate, glycolaldehyde as well as glyceraldehyde were reported (turnover number 8.6, 5.3 and 4.8 s−1, respectively). This enzyme groups outside the GAPN cluster as the recently characterized archaeal glyceraldehyde dehydrogenases of T. acidophilum (TA0809, Jung and Lee 2006; Reher and Schönheit 2006) and P. torridus (PTO0332, Reher and Schönheit 2006) as well as the so far uncharacterized S. solfataricus paralog (SSO1218) for which methylmalonate-semialdehyde dehydrogenase (acylating) is predicted (EC 1.2.1.27) (Fig. 5). Glyceraldehyde dehydrogenase is a constituent of the npED pathway, and for both characterized enzymes remote activity with GAP and glycolaldehyde has been reported. Thus, all characterized enzymes outside the GAPN cluster exhibit activity on glyceraldehyde and glycolaldehyde, although to a different extent (Brouns et al. 2006; Jung and Lee 2006; Reher and Schönheit 2006).

Phylogenetic tree based on an alignment of archaeal GAPN orthologs and Sulfolobus solfataricus GAPN paralogs. Sso-GAPN is indicated in bold and nodes supported by a bootstrap values >70% are indicated by black dots. Proteins for which the crystal structure has been resolved are underlined. Enzymes for which GAPN (non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase), glyceraldehyde dehydrogenase (GADH) and 2,5-dioxopentanoate dehydrogenase (DpoDH) have been reported are indicated with a black, gray and white arrow, respectively. Genes are indicated by their systematic gene names. Species abbreviations: Ape: Aeropyrum pernix; Pfu: Pyrococcus furiosus; Pto: Picrophilus torridus; Sac: Sulfolobus acidocaldarius; Smu: Streptococcus mutans; Sso: S. solfataricus; Sto: S. tokodaii; Tko: Thermococcus kodakaraensis; Tac: Thermoplasma acidophilum, Ttx: T. tenax; Hsp: Halobacterium sp. NRC-1
The close examination of the residues that constitute the active site and binding pockets of the co-substrate and the activators in Ttx-GAPN (Fig. 4) suggests that the currently uncharacterized members of the aldehyde dehydrogenase superfamily also differ in substrate specificity and/or regulatory properties. While all these residues, except for two residues involved in co-substrate binding (see above), are completely conserved in Sso-GAPN, only part of the active site residues and almost none of the residues involved in activator binding are conserved in the other S. solfataricus paralogs (Fig. 4). However, respective biochemical analyses have to be awaited in order to confirm these predictions.
In summary, GAPN homologs were identified in several archaeal genomes—preferentially hyperhermophiles—and phylogenetic analyses revealed five different paralogs of the aldehyde dhydrogenase superfamily in S. solfataricus of which only two—GAPN and 2,5-dioxopentanoate dehydrogenase—are characterized so far.
Physiological implications from the distribution of GAP converting enzymes in Archaea
Archaeal glycolytic pathways feature several modifications compared to analogous bacterial and eukaryal pathways. At the level of GAP oxidation, three alternative enzymes have been characterized: the ubiquitous GAPDH/PGK couple, and the non-phosphorylating archaeal counterparts GAPOR and GAPN (Table 2). Detailed enzymatic characterization as well as transcription analysis in P. furiosus and T. tenax (Brunner et al. 1998, 2001; Lorentzen et al. 2004; Schäfer and Schönheit 1993; Schut et al. 2003; Van der Oost et al. 1998) whose genomes encode all three GAP converting enzymes (Table 2) revealed a catabolic role for GAPN and GAPOR and an anabolic role for the GAPDH/PGK couple. These findings have been confirmed by a recent, excellent mutational approach in the hyperthermophilic anaerobe Thermococcus kodakarensis, which harbors all three GAP converting enzymes (Matsubara et al. 2006).
The physiological basis for this substitution is still unclear and a role in metabolic thermoadaptation has been suggested for the first time for Ttx-GAPN (Brunner et al. 2001). The archaeal non-phosphorylating GAPN/GAPOR by-pass the production of the extremely thermolabile intermediate 1,3-biphosphoglycerate (1,3-BGP), which has a half-life time of less than 2 min at 60°C, in the anabolic GAPDH/PGK reaction. Interestingly, an analysis of the distribution of genes encoding GAP converting enzymes seems to support this hypothesis (Table 2). Although GAPDH is present in all Archaea, GAPN and/or GAPOR seem to be found in hyperthermophiles (saccharolytic Archaea and glycogen-forming Methanogens). There are only few inconsistencies: (1) the hyperthermophiles Methanopyrus kandleri and Archaeoglobus fulgidus DSM4304 rely solely on GAPDH/PGK for GAP conversion. However, both organisms are reported to exhibit no saccharolytic growth and therefore might not need the catabolic counterparts GAPN or GAPOR (Stetter 1988). This theory is supported by the presence of a GAPOR encoding gene in the starch degrading A. fulgidus strain 7324 and its absence in the sequenced genome of A. fulgidus DSM4304 (Labes and Schönheit 2001; Siebers and Schönheit 2005). (2) The genomes of the mesophilic Archaea Methanococcus maripaludis and Halobacterium NRC1 contain genes most likely encoding GAPOR and GAPN, respectively, in addition to the GAPDH/PGK encoding genes (Table 2). As thermoadaptation is clearly not an issue in this organism (optimal growth temperature of M. maripaludis is 35–40°C; Halobacterium sp. NRC-1 at 42°C), another—yet unknown—advantage for retaining the GAPN/GAPOR encoding gene might be present. Two GAPDH encoding genes were identified in the genomes of Methanosarcina species suggesting different metabolic functions. A similar situation is reported in Bacillus subtilis which contains two distinct GAPDHs, one acting in gluconeogenic direction and the other acting in glycolytic direction (Fillinger et al. 2000).
In order to address if this by-pass is generally found in (hyper)thermophiles the distribution of GAP converting enzymes in (hyper)thermophilic bacteria was studied. Phylogenetic analyses revealed several hitherto uncharacterized homologs of the glyceraldehyde dehydrogenase superfamily and glyceraldehyde ferredoxin oxidoreductase family in (hyper)thermophilic bacteria (e.g. Aquifex aeolicus, Thermotoga maritime, Thermus thermophilus) (data not shown). None of these candidates groups within the GAPN (Fig. 5) or GAPOR cluster with characterized enzyme activities. Therefore it is still an open question if this by-pass is also found in thermophilic bacteria and also the utilization of alternative strategies such as substrate channeling/tunneling in order to prevent degradation of thermolabile intermediates has to be taken into account.
In summary, the preferred distribution of GAPN/GAPOR in hyperthermophilic Archaea suggests a role in metabolic thermoadaptation. However, a final answer is still pending and this hypothesis needs further analysis. So far, nothing is known about metabolite levels in hyperthermophiles and Archaea in general. Furthermore, no studies about thermal stability of metabolites and intermediates under in vivo conditions have been performed, and possible alternative mechanisms like substrate channeling have not been addressed.
Abbreviations
- ED:
-
Entner–Doudoroff
- sp:
-
Semi-phosphorylative
- np:
-
Non-phosphorylative
- EMP:
-
Embden-Meyerhof-Parnas
- G1P:
-
Glucose 1-phosphate
- GAP:
-
Glyceraldehyde 3-phosphate
- GAPN:
-
Non-phosphorylating GAP dehydrogenase
- GAPDH:
-
GAP dehydrogenase
- GAPOR:
-
GAP oxidoreductase
- KDG:
-
2-Keto-3-deoxygluconate
- KDPG:
-
2-Keto-3-deoxy-6-phosphogluconate
References
Ahmed H, Ettema TJ, Tjaden B, Geerling AC, Van der Oost J, Siebers B (2005) The semi-phosphorylative Entner–Doudoroff pathway in hyperthermophilic Archaea: a re-evaluation. Biochem J 390:529–540
Boyd DA, Cvitkovitch DG, Hamilton IR (1995) Sequence, expression, and function of the gene for the nonphosphorylating, NADP-dependent glyceraldehyde-3-phosphate dehydrogenase of Streptococcus mutans. J Bacteriol 177:2622–2627
Brinkman AB, Bell SD, Lebbink RJ, De Vos WM, Van der Oost J (2002) The Sulfolobus solfataricus Lrp-like protein LysM regulates lysine biosynthesis in response to lysine availability. J Biol Chem 277:29537–29549
Brouns SJ, Walther J, Snijders AP, Van de Werken HJ, Willemen HL, Worm P, De Vos MG, Andersson A, Lundgren M, Mazon HF, Van den Heuvel RH, Nilsson P, Salmon L, De Vos WM, Wright PC, Bernander R, Van der Oost J (2006) Identification of the missing links in prokaryotic pentose oxidation pathways: evidence for enzyme recruitment. J Biol Chem 281:27378–27388
Brunner NA, Hensel R (2001) Nonphosphorylating glyceraldehyde-3-phosphate dehydrogenase from Thermoproteus tenax. Methods Enzymol 331:117–131
Brunner NA, Brinkmann H, Siebers B, Hensel R (1998) NAD+-dependent glyceraldehyde-3-phosphate dehydrogenase from Thermoproteus tenax. The first identified archaeal member of the aldehyde dehydrogenase superfamily is a glycolytic enzyme with unusual regulatory properties. J Biol Chem 273:6149–6156
Brunner NA, Siebers B, Hensel R (2001) Role of two different glyceraldehyde-3-phosphate dehydrogenases in controlling the reversible Embden-Meyerhof-Parnas pathway in Thermoproteus tenax: regulation on protein and transcript level. Extremophiles 5:101–109
Buchanan CL, Connaris H, Danson MJ, Reeve CD, Hough DW (1999) An extremely thermostable aldolase from Sulfolobus solfataricus with specificity for non-phosphorylated substrates. Biochem J 343:563–570
Cobessi D, Tete-Favier F, Marchal S, Azza S, Branlant G, Aubry A (1999) Apo and holo crystal structures of an NADP-dependent aldehyde dehydrogenase from Streptococcus mutans. J Mol Biol 290:161–173
Condo I, Ciammaruconi A, Benelli D, Ruggero D, Londei P (1999) Cis-acting signals controlling translational initiation in the thermophilic archaeon Sulfolobus solfataricus. Mol Microbiol 34:377–384
Corwin LM, Fanning GR (1968) Studies of parameters affecting the allosteric nature of phosphoenolpyruvate carboxylase of Escherichia coli. J Biol Chem 243:3517–3525
De Rosa M, Gambacorta A, Nicolaus B, Giardina P, Poerio E, Buonocore V (1984) Glucose metabolism in the extreme thermoacidophilic archaebacterium Sulfolobus solfataricus. Biochem J 224:407–414
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Fillinger S, Boschi-Muller S, Azza S, Dervyn E, Branlant G, Aymerich S (2000) Two glyceraldehyde-3-phosphate dehydrogenases with opposite physiological roles in a nonphotosynthetic bacterium. J Biol Chem 275:14031–14037
Giardina P, de Biasi MG, de Rosa M, Gambacorta A, Buonocore V (1986) Glucose dehydrogenase from the thermoacidophilic archaebacterium Sulfolobus solfataricus. Biochem J 239:517–522
Gotz F, Schleifer KH (1975) Purification and properties of a fructose-1,6-diphosphate activated L-lactate dehydrogenase from Staphylococcus epidermidis. Arch Microbiol 105:303–312
Grogan DW (1989) Phenotypic characterization of the archaebacterial genus Sulfolobus: comparison of five wild-type strains. J Bacteriol 171:6710–6719
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Sys Biol 52:696–704
Habenicht A (1997) The non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase: biochemistry, structure, occurrence and evolution. Biol Chem 378:1413–1419
Johnsen U, Selig M, Xavier KB, Santos H, Schönheit P (2001) Different glycolytic pathways for glucose and fructose in the halophilic archaeon Halococcus saccharolyticus. Arch Microbiol 175:52–61
Jones DT, Taylor WR, Thornton JM (1992) The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8:275–282
Jung JH, Lee SB (2006) Identification and characterization of Thermoplasma acidophilum glyceraldehyde dehydrogenase: a new class of NADP+-specific aldehyde dehydrogenase. Biochem J 397:131–138
Jung JH, Lee SB (2005) Identification and characterization of Thermoplasma acidophilum 2-keto-3-deoxy-d-gluconate kinase: a new class of sugar kinases. Biotechnol Bioprocess Eng 10:535–539
Kardinahl S, Schmidt CL, Hansen T, Anemuller S, Petersen A, Schäfer G (1999) The strict molybdate-dependence of glucose-degradation by the thermoacidophile Sulfolobus acidocaldarius reveals the first crenarchaeotic molybdenum containing enzyme—an aldehyde oxidoreductase. Eur J Biochem 260:540–548
Kehrer D, Ahmed H, Brinkmann H, Siebers B (2007) Glycerate kinase of the hyperthermophilic Archaeon Thermoproteus tenax: new insights in the phylogenetic distribution and physiological role of members of the three different glycerate kinase families (Submitted BMC genomics)
Kim S, Lee SB (2005) Identification and characterization of Sulfolobus solfataricus d-gluconate dehydratase: a key enzyme in the non-phosphorylated Entner–Doudoroff pathway. Biochem J 387:271–280
Labes A, Schönheit P (2001) Sugar utilization in the hyperthermophilic, sulfate-reducing archaeon Archaeoglobus fulgidus strain 7324: starch degradation to acetate and CO2 via a modified Embden-Meyerhof pathway and acetyl-CoA synthetase (ADP-forming) Arch Microbiol 176:329–338
Lamble HJ, Heyer NI, Bull SD, Hough DW, Danson MJ (2003) Metabolic pathway promiscuity in the Archaeon Sulfolobus solfataricus revealed by studies on glucose dehydrogenase and 2-keto-3-deoxygluconate aldolase. J Biol Chem 278:34066–34072
Lamble HJ, Milburn CC, Taylor GL, Hough DW, Danson MJ (2004) Gluconate dehydratase from the promiscuous Entner–Doudoroff pathway in Sulfolobus solfataricus. FEBS Lett 576:133–136
Lamble HJ, Theodossis A, Milburn CC, Taylor GL, Bull SD, Hough DW, Danson MJ (2005) Promiscuity in the part-phosphorylative Entner–Doudoroff pathway of the archaeon Sulfolobus solfataricus. FEBS Lett 579:6865–6869
LeJohn HB, Jackson S (1968) Allosteric interactions of a regulatory nicotinamide adenine dinucleotide-specific glutamate dehydrogenase from Blastocladiella: a molecular model for the enzyme. J Biol Chem 243:3447–3457
Lorentzen E, Hensel R, Knura T, Ahmed H, Pohl E (2004) Structural basis of allosteric regulation and substrate specificity of the non-phosphorylating glyceraldehyde 3-phosphate dehydrogenase from Thermoproteus tenax. J Mol Biol 341:815–828
Markowitz VM, Korzeniewski F, Palaniappan K, Szeto E, Werner G, Padki A, Zhao X, Dubchak I, Hugenholtz P, Anderson I, Lykidis A, Mavromati K, Ivanova N, Kyrpides NC (2006) The integrated microbial genomes (IMG) system. Nucleic Acids Res 34:D344–D348
Matsubara K, Atomi H, Imanaka T (2006) A genetic analysis on enzymes involved in the conversion of glyceraldehyde 3-phosphate in the hyperthermophilic archaeon, Thermococcus kodakaraensis. Poster contribution. In: The 6th International Congress on Extremophiles, Brest, France
Milburn CC, Lamble HJ, Theodossis A, Bull SD, Hough DW, Danson MJ, Taylor GL (2006) The structural basis of substrate promiscuity in glucose dehydrogenase from the hyperthermophilic archaeon Sulfolobus solfataricus. J Biol Chem 281:14796–14804
Mukund S, Adams MW (1991) The novel tungsten-iron-sulfur protein of the hyperthermophilic archaebacterium, Pyrococcus furiosus, is an aldehyde ferredoxin oxidoreductase. Evidence for its participation in a unique glycolytic pathway. J Biol Chem 266:14208–14216
Mukund S, Adams MW (1995) Glyceraldehyde-3-phosphate ferredoxin oxidoreductase, a novel tungsten-containing enzyme with a potential glycolytic role in the hyperthermophilic archaeon Pyrococcus furiosus. J Biol Chem 270:8389–8392
Perozich J, Kuo I, Wang BC, Boesch J S, Lindahl R, Hempel J (2000) Shifting the NAD/NADP preference in class 3 aldehyde dehydrogenase. Eur J Biochem 267:6197–6203
Pohl E, Brunner N, Wilmanns M, Hensel R (2002) The crystal structure of the allosteric non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase from the hyperthermophilic archaeum Thermoproteus tenax. J Biol Chem 277:19938–19945
Reher M, Schönheit P (2006) Glyceraldehyde dehydrogenases from the thermoacidophilic euryarchaeota Picrophilus torridus and Thermoplasma acidophilum, key enzymes of the non-phosphorylative Entner–Doudoroff pathway, constitute a novel enzyme family within the aldehyde dehydrogenase superfamily. FEBS Lett 580:1198–1204
Reher M, Bott M, Schönheit P (2006) Characterization of glycerate kinase (2-phosphoglycerate forming), a key enzyme of the nonphosphorylative Entner–Doudoroff pathway, from the thermoacidophilic euryarchaeon Picrophilus torridus. FEMS Microbiol Lett 259:113–119
Ronimus RS, Morgan HW (2002) Distribution and phylogenies of enzymes of the Embden-Meyerhof-Parnas pathway from Archaea and hyperthermophilic bacteria support a gluconeogenic origin of metabolism. Archaea 1:199–221
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning—a laboratory manual, 2nd edn. Cold Spring Harbour Laboratory Press, New York
Schäfer T, Schönheit P (1993) Gluconeogensis from pyruvate in the hyperthermophilic archaeon Pyrococcus furiosus-involvement of reactions of the Embden-Meyerhof pathway. Arch Microbiol 159:254–363
Schicho RN, Snowden LJ, Mukund S, Park JB, Adams MW, Kelly RM (1993) Influence of tungsten on metabolic patterns in Pyrococcus furiosus, a hyperthermophilic Archaeon. Arch Microbiol 159:380–385
Schut GJ, Brehm SD, Datta S, Adams MW (2003) Whole-genome DNA microarray analysis of a hyperthermophile and an archaeon: Pyrococcus furiosus grown on carbohydrates or peptides. J Bacteriol 185:3935–3947
Schramm A, Siebers B, Tjaden B, Brinkmann H, Hensel R (2000) Pyruvate kinase of the hyperthermophilic crenarchaeote Thermoproteus tenax: physiological role and phylogenetic aspects. J Bacteriol 182:2001–2009
Selig M, Schönheit P (1994) Oxidation of organic-compounds to CO2 with sulfur or thiosulfate as electron-acceptor in the anaerobic hyperthermophilic Archaea Thermoproteus tenax and Pyrobaculum islandicum proceeds via the citric-acid cycle. Arch Microbiol 162:286–294
Selig M, Xavier KB, Santos H, Schönheit P (1997) Comparative analysis of Embden-Meyerhof and Entner–Doudoroff glycolytic pathways in hyperthermophilic Archaea and the bacterium Thermotoga. Arch Microbiol 167:217–232
She Q, Singh RK, Confalonieri F, Zivanovic Y, Allard G, Awayez MJ, Chan-Weiher CC, Clausen IG, Curtis BA, De Moors A, Erauso G, Fletcher C, Gordon PM, Heikamp-de Jong I, Jeffries AC, Kozera CJ, Medina N, Peng X, Thi-Ngoc HP, Redder P, Schenk ME, Theriault C, Tolstrup N, Charlebois RL, Doolittle WF, Duguet M, Gaasterland T, Garrett RA, Ragan MA, Sensen CW, Van der Oost J (2001) The complete genome of the crenarchaeon Sulfolobus solfataricus P2. Proc Natl Acad Sci USA 98:7835–7840
Siebers B, Hensel R (1993) Glucose catabolism of the hyperthermophilic Aarchaeum Thermoproteus tenax. FEMS Microbiol Lett 111:1–8
Siebers B, Schönheit P (2005) Unusual pathways and enzymes of central carbohydrate metabolism in Archaea. Curr Opin Microbiol 8:695–705
Siebers B, Wendisch VF, Hensel R (1997) Carbohydrate metabolism in Thermoproteus tenax: in vivo utilization of the non-phosphorylative Entner–Doudoroff pathway and characterization of its first enzyme, glucose dehydrogenase. Arch Microbiol 168:120–127
Siebers B, Brinkmann H, Dörr C, Tjaden B, Lilie H, Van der Oost J, Verhees CH (2001) Archaeal fructose-1,6-bisphosphate aldolases constitute a new family of archaeal type class I aldolase. J Biol Chem 276:28710–28718
Siebers B, Tjaden B, Michalke K, Dörr C, Ahmed H, Zaparty M, Gordon P, Sensen CW, Zibat A, Klenk HP, Schuster SC, Hensel R (2004) Reconstruction of the central carbohydrate metabolism of Thermoproteus tenax by use of genomic and biochemical data. J Bacteriol 186:2179–2194
Slupska MM, King AG, Fitz-Gibbon S, Besemer J, Borodovsky M, Miller JH (2001) Leaderless transcripts of the crenarchaeal hyperthermophile Pyrobaculum aerophilum. J Mol Biol 309:347–360
Snijders AP, Walther J, Peter S, Kinnman I, De Vos MG, Van de Werken HJ, Brouns SJ, Van der Oost J, Wright PC (2006) Reconstruction of central carbon metabolism in Sulfolobus solfataricus using a two-dimensional gel electrophoresis map, stable isotope labelling and DNA microarray analysis. Proteomics 6:1518–1529
Stetter KO (1988) Archaeoglobus fulgidus gen. nov., sp. nov.: a new taxon of extremely thermophilic Archaebacteria. Sys App Microbiol 10:172–173
Theodossis A, Walden H, Westwick EJ, Connaris H, Lamble HJ, Hough DW, Danson MJ, Taylor GL (2004) The structural basis for substrate promiscuity in 2-keto-3-deoxygluconate aldolase from the Entner–Doudoroff pathway in Sulfolobus solfataricus. J Biol Chem 279:43886–43892
Tolstrup N, Sensen CW, Garrett RA, Clausen IG (2000) Two different and highly organized mechanisms of translation initiation in the archaeon Sulfolobus solfataricus. Extremophiles 4:175–179
Tomlinson GA, Koch TK, Hochstein LI (1974) The metabolism of carbohydrates by extremely halophilic bacteria: glucose metabolism via a modified Entner–Doudoroff pathway. Can J Microbiol 20:1085–1091
Van der Oost J, Schut G, Kengen SW, Hagen WR, Thomm M, De Vos WM (1998) The ferredoxin-dependent conversion of glyceraldehyde-3-phosphate in the hyperthermophilic archaeon Pyrococcus furiosus represents a novel site of glycolytic regulation. J Biol Chem 273:28149–28154
Verhees CH, Kengen SWM, Tuininga JE, Schut GJ, Adams MW, De Vos WM, Van der Oost J (2003) The unique features of glycolytic pathways in Archaea. Biochem J 375:231–246; errata notification:(2004) Biochem J 377:819–822
Yoshida A, Rzhetsky A, Hsu LC, Chang C (1998) Human aldehyde dehydrogenase gene family. Eur J Biochem 251:549–557
Zillig W, Stetter KO, Wunderl S, Wunderl S, Priess H, Scholz J (1980) The Sulfolobus-”Caldariella” group: taxonomy on the basis of the structure of DNA-dependent RNA polymerases. Arch Microbiol 125:259–269
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (SPP 1112, SI642/6-1) and by a Rubicon grant to T.E. from the Netherlands Organisation for Scientific Research (NWO).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by D.A. Cowan.
Thijs J. G. Ettema and Hatim Ahmed have contributed equally to this work.
Rights and permissions
About this article
Cite this article
Ettema, T.J.G., Ahmed, H., Geerling, A.C.M. et al. The non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase (GAPN) of Sulfolobus solfataricus: a key-enzyme of the semi-phosphorylative branch of the Entner–Doudoroff pathway. Extremophiles 12, 75–88 (2008). https://doi.org/10.1007/s00792-007-0082-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-007-0082-1