
Abstract
The organometallic H-cluster at the active site of the [FeFe]-hydrogenase serves as the site of reversible binding and reduction of protons to produce H2. The H-cluster is unique in biology, and consists of a 2Fe subcluster tethered to a typical [4Fe–4S] cluster by a single cysteine ligand. The remaining ligands to the 2Fe subcluster include three carbon monoxides, two cyanides, and a dithiomethylamine. This mini-review will focus on the significant advances in recent years in understanding the pathway for H-cluster biosynthesis, as well as the structures, roles, and mechanisms of the three enzymes directly involved.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The [FeFe]- and [NiFe]-hydrogenases catalyze reversible hydrogen oxidation and both require π-acid nonprotein ligands at the active site metal cluster for catalysis [1–3]. The [FeFe]-hydrogenases, the focus of this review, have one bridging and two terminal carbon monoxides, two terminal cyanides, and a bridging dithiomethylamine all coordinating a 2Fe cluster that is bridged to a [4Fe–4S] cluster (Fig. 1) [4–9]. The iron distal to the [4Fe–4S] cluster is the presumed site of reversible proton reduction, and the π-acid ligands are thought to stabilize low oxidation states of the iron ions while reducing back-donation to H2 to prevent hydride formation [10, 11]. The dithiomethylamine ligand bridges the two irons in the subcluster and is proposed to be involved in proton transfer during catalysis [12].

The structure of the [FeFe]-hydrogenase (CpI) from Clostridium pasteurianum. Color scheme: gray active site domain containing the H-cluster; deep purple domain 2 containing two [4Fe–4S] clusters; purple domain 3 containing one [4Fe–4S] cluster; light pink domain 4 containing one [2Fe–2S] cluster. Atoms are colored: gray C; blue N; red O; rust Fe; yellow S. A series of iron–sulfur clusters, presumably involved in shuttling electrons from protein partners, are present near the active site H-cluster where protons are reduced to form H2. (PDB ID: 3C8Y)
Production of an active [FeFe]-hydrogenase requires simultaneous expression of hydA (the structural gene) and three additional genes, hydE, hydF, and hydG [13, 14]. Deduced amino acid sequences indicate that two of the accessory proteins, HydE and HydG, belong to the radical S-adenosyl-l-methionine (SAM) superfamily [14] raising the intriguing question of how radical SAM chemistry is involved in H-cluster biogenesis. The amino acid sequence of the third accessory protein, HydF, contains a grouping of potential metal-binding residues in addition to Walker A P-loop and Walker B Mg2+ binding motifs characteristic of NTPases [14]. In the 10 years since the initial discovery of these accessory proteins, considerable progress has been made in understanding the roles of each enzyme in [FeFe]-hydrogenase maturation.
[FeFe]-hydrogenase structure and mechanism
The first published X-ray crystal structures of the [FeFe]-hydrogenases from Clostridium pasteurianum (CpI) [4] and Desulfovibrio desulfuricans (Dd) [5] revealed the unique architecture of the 6Fe active site (Fig. 1). While the details of the diatomic ligands would only be revealed by later studies [15], these structures pointed to the unusual nature of the cluster where protons are reduced to form H2. Four of the six irons are associated with a [4Fe–4S] cubane similar to those found widely in biology, with the cubane bridged by one of its thiolate ligands to a 2Fe subcluster that has no additional attachment to the protein. The identities of the diatomic ligands of the 2Fe subcluster were determined via FTIR spectroscopic studies [15]. The precise nature of the bridging dithiolate, specifically the identity of the bridgehead group, has eluded direct determination, although multiple studies have now provided strong support for dithiomethylamine [6, 8, 9]. The unusual structure of this 2Fe subcluster has inspired numerous elegant synthetic approaches to modeling both the structure and the reactivity of the H-cluster [16].
The remaining iron–sulfur clusters seen in the CpI and Dd structures are bound to domains that are distinct from the active site domain, and likely serve as conduits for electron transfer from in vivo protein partners to the active site during hydrogen production. The quantity of these accessory iron–sulfur clusters vary widely. The [FeFe]-hydrogenase from Chlamydomonas reinhardtii (Cr) lacks any accessory clusters, while CpI contains four additional clusters. The relative structural simplicity of the Cr enzyme has been exploited for investigating maturation of the H-cluster, as will be detailed later in this review.
The mechanism for reversible proton reduction by the [FeFe]-hydrogenase is under active investigation, and a recent proposal put forth by Lubitz et al. [17], and supported by Mulder et al. [18], is discussed here. In this mechanism, the oxidized (Hox) state containing a [4Fe–4S]2+ cluster bridged to the FeIFeII subcluster is reduced to the Hred state ([4Fe–4S]2+–FeIFeI) concomitant with protonation of the bridgehead nitrogen to the dialkylammonium state (Fig. 2). Further reduction of this Hred state to the super-reduced state (Hsred, [4Fe–4S]+–FeIFeI) is accompanied by migration of a proton from the bridgehead group to a site adjacent to the 2Fe subcluster distal iron (Fed). In the next step of the proposed mechanism, this proton undergoes oxidative addition at the Fed of the 2Fe subcluster to form a [4Fe–4S]2+–FeIFeII–H− species with concomitant protonation of the bridgehead nitrogen. Donation of the bridgehead proton to the hydride produces a bound dihydrogen species which can then dissociate to regenerate the initial Hox state. As depicted in Fig. 2, changes in coordination of the bridging CO during the catalytic cycle are also proposed.

Proposed mechanism for reversible proton reduction catalyzed by the [FeFe]-hydrogenase. Key chemical changes relative to the Hox state are highlighted in orange
The key requirements of catalysis for the [FeFe]-hydrogenase therefore appear to be (1) a 2Fe subcluster that can achieve the low oxidation states required for efficient reversible H2 oxidation; (2) an electronically coupled [4Fe–4S] cluster that can accommodate a reducing equivalent in Hsred such that oxidative addition of a proton to the distal iron can occur; and (3) an appropriately positioned base (the bridgehead amine) that can serve as a conduit for protons to the distal Fe of the H-cluster. It is clear that the unusual 2Fe subcluster of the H-cluster is exquisitely structurally tuned for catalysis, and that its nonprotein ligands play central roles in defining the reactivity at this active site [11].
Early clues to the maturation process
In 2004, Posewitz and coworkers [14] first identified genes for three accessory hydrogenase proteins, HydE, HydF, and HydG, required for maturation of an active [FeFe]-hydrogenase in Cr. The authors analyzed available genomes and found organisms containing an [FeFe]-hydrogenase also had the hydE, hydF, and hydG genes. The work also provided the first example of heterologous expression of an active [FeFe]-hydrogenase, by co-expression of Cr hydEF, hydG, and hydA (encoding the hydrogenase) genes in Escherichia coli [14]. A subsequent study utilized co-expression of the hydE, hydF, and hydG genes from Clostridium acetobutylicum (Ca) with hydA genes from several organisms, resulting in production of an active hydrogenase [19]. Site-directed mutagenesis revealed that the radical SAM cysteine motifs of both HydE and HydG, and the GTPase domain and putative cluster-binding motif of HydF, were all essential to maturation, thereby providing important guiding principles to future maturation studies [19].
The first in vitro activation of [FeFe]-hydrogenase was accomplished by mixing an E. coli cell extract in which Ca HydE, HydF, and HydG were co-expressed with an E. coli cell extract in which HydA was expressed [20]. Extract of the maturase strain that was heat treated or filtered to remove protein was incapable of activating HydA, suggesting that the activation process involved direct interaction between one or more of the maturation proteins and HydA. Further, the activation of HydA by the maturase cell extract did not require the addition of potential cluster precursors, suggesting that a preformed component synthesized by the maturation machinery was transferred from one of the maturases to HydA to effect activation [20].
Boyer et al. [21] reported a cell-free system for the synthesis and maturation of [FeFe]-hydrogenase. Their system utilized the Hyd maturases from Shewanella oneidensis (So) expressed in E. coli, and they were able to produce and activate hydrogenases from a variety of sources using these E. coli extracts, with activation enhanced by addition of iron and sulfide [21]. Further studies of this in vitro maturation system demonstrated that the addition of iron, sulfide, SAM, tyrosine, and cysteine to maturation reactions containing purified HydA and dialyzed E. coli extract containing the maturation proteins provided a fivefold enhancement in hydrogenase activity relative to the unsupplemented maturation reaction [22]. The added iron and sulfide likely reconstituted iron–sulfur clusters in the component proteins, while the added SAM would be expected to be essential for the function of the radical SAM proteins HydE and HydG. It was speculated that the enhancement provided by tyrosine and cysteine pointed to their roles as substrates for H-cluster biosynthesis [22].
The cumulative evidence from these early studies therefore supported the idea that all three maturation proteins, HydE, HydF, and HydG, were required for biosynthesis of the H-cluster. They also supported a role for radical SAM chemistry in the process, and for the potential involvement of tyrosine and cysteine as substrates.
The nature of inactive HydA
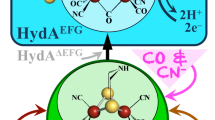
Important clues to the roles of the maturase enzymes came from characterization of HydA expressed in the absence of maturases (HydAΔEFG). HydAΔEFG was inactive presumably due to the lack of a functional H-cluster; however, it was unclear whether HydAΔEFG was completely devoid of metals in its H-cluster binding pocket, or whether it contained a partial or full [4Fe–4S] cluster but no 2Fe subcluster, or a 6Fe cluster lacking the appropriate nonprotein ligands. The answer was revealed through detailed characterization of HydAΔEFG using spectroscopic and structural studies. Purified Cr HydA expressed in E. coli without the maturation enzymes showed UV–visible, EPR, EXAFS, and Mössbauer spectroscopic properties characteristic of a [4Fe–4S] cluster [23]. HydAΔEFG containing this [4Fe–4S] cluster was activated upon addition of an E. coli extract containing the three maturation proteins HydE, HydF, and HydG. However, if the [4Fe–4S] cluster was removed, no activation occurred under the same experimental conditions [23]. These studies provided important insights into the maturation process, demonstrating that maturation required a preformed [4Fe–4S] cluster in the active site of HydA, and therefore implicating the maturases in synthesizing only the 2Fe subcluster.
Structural characterization of the inactive Cr HydAΔEFG revealed the presence of a [4Fe–4S] cluster in the H-cluster binding pocket, with an open cavity where the 2Fe subcluster normally resides (Fig. 3) [24]. It was inferred that H-cluster assembly and hydrogenase activation occur in a stepwise fashion, with the [4Fe–4S] cluster synthesized by the general iron–sulfur cluster assembly machinery and the 2Fe subcluster synthesized and inserted by the Hyd-specific maturases [24]. A channel lined with positive charge leads into the H-cluster binding pocket, and comparison of the inactive HydAΔEFG structure to the CpI structure indicates this channel closes upon maturation due to the conformational changes in two conserved loops (Fig. 3). Further evidence for stepwise H-cluster assembly was provided by nuclear resonance vibrational and EPR spectroscopic studies of HydAΔEFG matured with 57Fe-enriched maturase extract; the results clearly indicate 57Fe present only in the 2Fe subcluster of the H-cluster [25].

Structural changes in HydA upon 2Fe subcluster insertion. The X-ray crystal structure of Cr HydA expressed in the absence of the maturase enzymes (left, PDB ID: 3LX4) reveals an active site containing a preformed [4Fe–4S] cluster. The fully mature CpI enzyme (right, PDB ID: 3C8Y) contains the full 6Fe H-cluster, with the 2Fe subcluster synthesized and inserted via the action of the maturation enzymes HydE, HydF, and HydG. The accessory FeS cluster domains in the CpI enzyme are omitted for clarity. Two ß-strands in both structures are colored purple to emphasize structural similarities; the loop region that moves during 2Fe subcluster insertion is highlighted in blue. (Color scheme: C gray; N blue; O red; S yellow; Fe rust; undefined magenta) Adapted from [24]
These studies on the inactive and mature forms of HydA provided a critical framework for understanding the roles of the maturation enzymes: clearly demonstrating HydE, HydG, and HydF’s requirement for the synthesis of the 2Fe subcluster of the H-cluster, complete with unusual nonprotein ligands, and its delivery to HydA that already contains the [4Fe–4S] portion of the H-cluster.
HydF is likely a scaffold/carrier for the 2Fe subcluster
Iron–sulfur clusters on HydF
HydF binds iron–sulfur clusters, although the precise nature of the clusters has been a matter of some debate [26–30]. Thermotoga maritima (Tm) HydF expressed in E. coli appears to bind an unusual [4Fe–4S] cluster coordinated by three cysteines and a fourth, exchangeable ligand [26]. Ca HydF expressed alone in E. coli exhibits UV–visible and EPR spectroscopic properties that have been interpreted as arising from a mixture of [4Fe–4S] and [2Fe–2S] clusters [27, 28]; however, others have questioned the assignment of the [2Fe–2S] EPR signal, suggesting that it may instead arise from a [3Fe–4S]+ cluster or an organic radical [29, 30].
Interestingly, our studies of Ca HydFEG (purified from E. coli that co-expressed HydE and HydG) exhibit only the [4Fe–4S]+ signal in the reduced state [28]. Further, FTIR spectroscopy of Ca HydFEG expressed either heterologously in E. coli [28] or homologously in Ca [31] provided clear evidence for the presence of metal-coordinated CO and CN− (Fig. 4). No such diatomics were observed in FTIR spectra of HydFΔEG [28]. At the time, we proposed that a [2Fe–2S] cluster on HydF underwent modification by the radical SAM chemistry of HydE and HydG to generate an H-cluster precursor containing CO and CN− [28]. Given the debate regarding whether a [2Fe–2S] cluster is present in HydFΔEG, other possibilities need to be considered. One intriguing alternative, that the radical SAM enzyme HydG provides both the iron and the diatomics to assemble a 2Fe precursor on HydF, can be inferred from recent studies that will be discussed further in the section on HydG.

HydF expressed heterologously (top, Shepard et al.) or homologously (bottom, Czech et al., A is dithionite-reduced, while B is in the absence of dithionite) in the presence of HydE and HydG contains Fe–CN and Fe–CO species, as evidenced by FTIR spectroscopy. Reprinted with permission from [28] (top) and [31] (bottom)
Further evidence that HydF serves as a scaffold/carrier for an H-cluster precursor was provided by EXAFS studies of HydFEG, which suggested that the 2Fe subcluster was linked to the [4Fe–4S] cluster on HydF in a manner reminiscent of the H-cluster on HydA [32]. Interestingly, synthetic 2Fe subclusters can also be loaded onto HydF and delivered to HydA to produce an active enzyme; in the case of these synthetic clusters, it appears that one of the cyanide ligands may bridge the 2Fe subcluster to the [4Fe–4S] cluster on HydF [8].
GTP binding and hydrolysis
HydF is a cation-activated GTPase, but the specific role that GTP binding and hydrolysis play in the maturation process is unresolved [28]. Activation of HydAΔEFG by HydFEG has been shown to be unaffected by the presence of GTP, suggesting that GTP binding/hydrolysis plays a role in the earlier steps in maturation, perhaps in the interaction between HydF and the other two maturases. Consistent with this hypothesis, the presence of HydE or HydG increases the rate of HydF-catalyzed GTP hydrolysis by ~50 % [28]. Further, addition of GTP increases dissociation rates of the radical SAM maturases in both HydE–HydF and HydG–HydF complexes [33]. Delineating the precise role for GTP binding and hydrolysis, however, awaits further studies.
Insights from the structure of HydF
An X-ray crystal structure of the apo-form of Thermotoga neapolitana (Tn) HydF has provided a structural framework for evaluating its function and mechanism (Fig. 5) [34]. The structure reveals a multimeric protein with each subunit having an N-terminal GTPase domain, a central dimerization domain, and a C-terminal iron–sulfur cluster-binding domain. A tetrameric structure is evident, composed of dimeric units in which two subunits have more extensive contacts. Although the structure is devoid of iron–sulfur clusters, it reveals a spatial grouping of the conserved putative iron–sulfur cluster-binding ligands (CxHx46–53HCGGC) previously identified by Posewitz et al. [14], and shown to be functionally important by King et al. [19]. Site-directed mutagenesis of HydF from two different sources (Ca and Tn), combined with EPR and HYSCORE characterization, suggested that the three cysteines in the metal-binding motif are important for iron–sulfur cluster coordination, while the roles of the two histidines are more ambiguous, with H352 coordinating only in the Ca protein [30]. Additional site-directed mutagenesis studies of the Ca protein, coupled with EPR spectroscopic characterization, implicated all three cysteines together with H306 in cluster binding [35]. Significantly decreased iron content was observed upon any substitution at C304 and C356, while at C353 and H306, only substitutions that completely removed the possibility of metal coordination (i.e., substitutions to A) resulted in low iron content. All variants at the C positions were inactive in in vitro HydA activation assays. The variants at H306 displayed dramatically decreased but detectable activity, particularly H306Q (13 % of wild type). All variants that had iron content sufficient for EPR characterization showed EPR spectra similar to that observed for wild-type HydF.

The maturase HydF has GTPase and FeS domains. Maturase HydF from Thermotoga neapolitana is depicted (left) in dimer form with residues in the GTPase and FeS binding domains highlighted. One subunit is illustrated in blue, while the other subunit is shown in gray. The GTPase Domain: housed in this domain, the residues associated with GTP binding and hydrolysis are highlighted in cyan for distal GTP specificity (NKxD), blue for the G-5 loop domain, purple for the Walker A P-loop (GxxNxGKS) and magenta for the Walker B Mg2+ binding loop (LxDTxG). This magnified box view is rotated 90° counter clock-wise in the xy plane from the full dimer structure on the left. The FeS Binding Domain: the three cysteine residues highlighted are the only cysteine residues in the monomer. (Color scheme in expanded view: N blue; O red; S yellow; C, same color as highlighted stick) (PDB ID: 3QQ5)
Function of HydF
While it is clear that HydF binds iron–sulfur clusters, the precise nature of these clusters, how they are bound, and even their role in maturation are still unclear. The cluster-binding motif has only three cysteines and two histidines; while this is a sufficient number of ligands to coordinate a single [4Fe–4S] cluster, coordination of both a [4Fe–4S] and a [2Fe–2S] cluster would require additional amino acid residues, or coordination of the clusters across subunit interfaces where residues from two different subunits are utilized. A bound [2Fe–2S] cluster could be modified by HydE and HydG to synthesize a CO, CN− and dithiolate ligated 2Fe subcluster. Alternatively, a bound [2Fe–2S] cluster could be displaced by Fe–CO–CN units delivered by HydG, or a vacant cavity might receive Fe–CO–CN units. Regardless of these alternative pathways, considerable evidence from multiple labs points to the assembly of a 6Fe precursor that closely resembles the H-cluster on HydF. Although some recent work suggests that HydF’s role may be optional in hydrogenase maturation [29], this interpretation is contrary to the early in vivo studies of King et al. and Posewitz et al. [13, 14, 19], as well as more recent in vitro studies which all point to an indispensable role for HydF [20, 27, 31].
HydG: radical-mediated CO and CN− synthesis
Reaction catalyzed by HydG
The first clue to the substrate for HydG was reported in 2009, when Pilet et al. [36] showed that HydG catalyzed the Cα–Cβ bond cleavage of tyrosine to produce p-cresol in a SAM-dependent reaction. Subsequently, we showed that the remaining products of HydG-catalyzed tyrosine cleavage are CN− and CO (Fig. 6) [37, 38]. CN− was detected by acid denaturation of the protein followed by derivatization [37], while CO was detected during the assay by including deoxyhemoglobin and monitoring its conversion to carboxyhemoglobin [38]. In both sets of experiments, uniformly 13C-labeled tyrosine was used to demonstrate that these diatomics were derived from tyrosine. A subsequent report by Kuchenreuther et al. [39] utilized labeled tyrosine in a cell-free system to show that all of the diatomic ligands on activated [FeFe]-hydrogenase are derived from tyrosine. Together, these studies reveal that HydG utilizes tyrosine as a substrate in the synthesis of CO and CN−, and these diatomics are ultimately delivered to the hydrogenase to assemble the H-cluster. The multiple lines of evidence for the presence of a CO and CN– ligated 2Fe species on HydF after its co-expression with HydE and HydG provides strong evidence the diatomics produced by HydG are first transferred to HydF, either as free ligands or as Fe-bound species prior to HydA maturation [28, 31, 32].

HydG iron–sulfur clusters (top) and reaction catalyzed (bottom). HydG binds two distinct iron–sulfur clusters: the radical SAM [4Fe–4S] cluster in the N-terminal domain (top left) and a second site-differentiated cluster in the C-terminal domain (top right). The radical SAM cluster is necessary and sufficient for reductive cleavage of SAM and tyrosine Cα–Cβ bond cleavage to produce p-cresol, while the C-terminal cluster is essential for CO production [40]
The iron–sulfur clusters of HydG and their roles in catalysis
HydG binds two [4Fe–4S] clusters, a radical SAM cluster at a CX3CX2C motif in the N-terminal domain of the protein, and an additional cluster at a CX2CX22C motif in the C-terminal domain (Fig. 6); [38, 40] both clusters are essential for hydrogenase maturation [19]. The clusters are both site-differentiated, as indicated by the presence of only three cysteines in the cluster-binding motifs. Radical SAM clusters utilize the unique iron site to bind SAM via the amino and carboxylate groups in a classical 5-member chelate ring [41–43]. The mechanistic significance, if any, of site differentiation at the C-terminal cluster in HydG is unresolved. Each cluster has distinct EPR spectroscopic properties, and only the N-terminal cluster interacts directly with SAM [38, 40]. By utilizing a series of HydG proteins in which either cluster-binding residues were changed to serine or alanine, or in which entire cluster-binding domains were deleted, the roles for the two individual clusters in HydG have been delineated [40, 44]. The N-terminal cluster is absolutely required for the reductive cleavage of SAM and the cleavage of tyrosine to produce p-cresol [40], and both of these activities occur independently of the presence or absence of the C-terminal cluster. However, production of the diatomic products by HydG absolutely requires the presence of the C-terminal domain [40]. Substitution of one of the cluster-binding cysteines in the C-terminal motif completely abolishes CO formation, although low levels of CN− are still detected with this variant [40]. Importantly, in variants where diatomic ligand production is turned off, increased levels of glyoxylate are detected; glyoxylate is the hydrolysis product of dehydroglycine, the latter which is the precursor to the diatomics [40].
The mechanism of HydG therefore involves reductive cleavage of SAM at the N-terminal cluster, with the resulting 5′-deoxyadenosyl radical (dAdo·) abstracting a hydrogen atom from tyrosine, and this tyrosyl radical undergoing Cα–Cβ bond cleavage. Recent evidence indicates that this bond cleavage is heterolytic, as rapid freeze-quench EPR spectroscopy has provided evidence for an intermediate p-cresol radical [45]. HYSCORE and ENDOR data during turnover in the presence of uniformly 13C and 15N-labeled tyrosine provide evidence for interaction of either tyrosine or a tyrosine-derived fragment with the C-terminal cluster [45]. Homology models of HydG predict that the two iron–sulfur clusters are more than 20 Å apart [44]; given the requirement for direct H-atom abstraction from tyrosine by dAdo· generated at the N-terminal cluster, it seems unlikely that tyrosine coordination to the C-terminal cluster would be catalytically relevant. Thus, we suspect that a tyrosine-derived fragment, likely dehydroglycine or a similar species, gives rise to the observed coupling [45].
Stopped flow FTIR studies have recently revealed the formation of [Fe(CO)CN−] and subsequently [Fe(CO)2CN−] species on HydG (Fig. 7) [46]. These intriguing species were proposed to form at the unique iron of the C-terminal cluster of HydG, followed by transfer to HydA either directly or via HydF as intermediary [46]. The implication is that the diatomics produced by HydG are transferred as metal-bound species rather than free diatomics, and thus that HydG provides both the diatomic ligands and the iron in the formation of the 2Fe subcluster of the H-cluster [46]. Evidence for the transfer of iron from HydG to the 2Fe subcluster of HydA was provided by 57Fe ENDOR spectroscopy [46]. Formation of iron–cyano-carbonyl species on HydG would be expected to diminish CO detection via deoxyhemoglobin binding, and therefore could explain why CO has been observed at <1:1 stoichiometry with CN− [38]. However, CO formation by purified Ca HydG occurs with burst phase kinetics at an initial rate comparable to p-cresol and CN− formation [38], which seems inconsistent with a mechanism producing iron-bound CO and CN− products via decomposition of dehydroglycine on the C-terminal cluster. For So HydG, time-resolved FTIR-monitored production of carboxymyoglobin (MbCO) revealed CO production only at longer times, which would be more consistent with formation of iron-carbonyl-cyano species [46]. Although significant advances have been made in recent years in understanding the remarkable reaction catalyzed by HydG, future work will further probe mechanistic details, and will likely illuminate a unified mechanism for HydG catalysis.

FTIR spectra of the two iron–cyano-carbonyl species generated on So HydG during catalytic turnover of tyrosine. Reprinted with permission from [45]
HydE: the enigmatic maturase
As with HydG, HydE was shown early on to bind iron–sulfur clusters and to catalyze the reductive cleavage of SAM, consistent with its assignment as a radical SAM enzyme [47]. Given the demonstration that HydG synthesizes CO and CN− from tyrosine [37, 38], it is presumed that HydE synthesizes the bridging dithiomethylamine of the H-cluster. Identification of a substrate for HydE, however, has not yet been reported. Clues to the substrate for HydE may lie in early work on the cell-free system, which showed that the presence of cysteine, tyrosine, and SAM enhanced hydrogenase activation [22]. Given tyrosine is now known to be the substrate of HydG, and SAM is required for radical SAM activity, cysteine seems a likely possibility as the substrate for HydE, although this has not been demonstrated. Further clues emerge from the high-resolution crystal structures of Tm HydE in the presence of S-adenosyl-l-homocysteine [48], SAM, and 5′-deoxyadenosine and methionine [49]. HydE exhibits a TIM barrel fold, with the radical SAM [4Fe–4S] cluster at the top of the barrel and coordinated at the unique iron by SAM (or SAH or Met) via the amino and carboxylate moieties (Fig. 8) [48]. An auxiliary cluster is also bound in solved HydE structures in varying iron–sulfur cluster composition; however, its position outside the TIM barrel together with the fact that its cluster-binding ligands are not conserved throughout all HydE sequences suggest that this second cluster is unlikely to have a critical role in catalysis [48, 50]. In silico docking studies with >20,000 small molecules revealed that the best hits were molecules with net neutral to negative charge, a carboxylate, and a partial positive charge at one end [48]. Further, crystal soaking experiments revealed that thiocyanate bound readily at a site previously occupied by chloride at the bottom of the barrel [48]. Altogether, the crystallographic characterization of HydE has provided an important framework for evaluating potential substrates.

The maturase HydE from Thermotoga maritima viewed from the side (a) and top (b) of the TIM barrel. The TIM barrel α-helices and β-strands are highlighted in pale green and blue–green, respectively. Note the location of the [2Fe–2S] cluster ~20 Å from the radical SAM cluster and outside the TIM barrel. Figure panel b is rotated 90° about the x-axis relative to panel a. Fig. 8a. (Color scheme: SAM blue; Fe rust; S yellow–orange.) (PDB ID: 3IIZ)
Putting it together: hypothetical pathways for maturation of [FeFe]-hydrogenase
The pathway by which Nature synthesizes the organometallic H-cluster at the active site of the [FeFe]-hydrogenase is being revealed through a variety of approaches that include investigation of maturation using purified enzymes or cell-free systems, defining the biochemical properties and reactions catalyzed by individual purified maturation enzymes, and utilizing spectroscopic and structural approaches to gain insight into the biosynthetic process. While the picture of [FeFe]-hydrogenase maturation is by no means complete, we present in Fig. 9 two reasonable pathways based on the available experimental data.

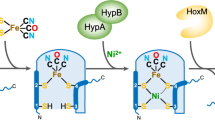
Two possible pathways for [FeFe]-hydrogenase maturation. In both pathways, the maturation process involves synthesis and assembly of the 2Fe subcluster of the H-cluster, followed by insertion of this 2Fe subcluster to generate the active hydrogenase. In pathway (a), a [2Fe–2S] cluster on HydF is modified by HydE as the latter synthesizes the dithiomethylamine bridging ligand. HydG then utilizes tyrosine to synthesize CO and CN−, which are delivered to HydF and coordinate the modified 2Fe cluster, prior to transfer of the 2Fe subcluster to HydA. In pathway (b), HydG synthesizes cyano-carbonyl–iron species upon degradation of tyrosine, and transfers these iron-bound diatomics to HydF, where a 2Fe subcluster precursor is assembled. HydE then synthesizes and delivers the dithiomethylamine ligand prior to 2Fe subcluster transfer to HydA. Asterisks indicate unknown ligands
Multiple lines of evidence support the idea that all the maturation proteins (HydE, HydF, and HydG) are required for the synthesis of the 2Fe subcluster of the H-cluster, with the [4Fe–4S] subcluster presumably being synthesized by the ISC and/or Suf iron–sulfur cluster assembly machinery [51]. In Fig. 9, we show two putative pathways by which the 2Fe subcluster is synthesized on HydF by radical SAM-mediated synthesis and delivery of the unique nonprotein ligands. In Fig. 9a, HydE modifies a [2Fe–2S] cluster on HydF by synthesis and insertion of the dithiomethylamine bridging ligand. Then, HydG catalyzes the formation of CO and CN−, which are delivered to the 2Fe subcluster. Direct interactions between the radical SAM maturases and HydF have been reported [27, 33], and may be important for efficient delivery of the products of HydE and HydG. The result is an H-cluster-like species on HydF, as has been supported by FTIR and EXAFS studies [28, 31, 32]. The assembled 2Fe subcluster is then transferred to HydA.
In Fig. 9b, we show an alternate pathway in which iron-bound cyanide and carbon monoxide are first synthesized on HydG, and then are delivered to HydF as cyano-carbonyl–Fe units, where dithiomethylamine addition stitches the two mononuclear Fe species together. The synthesis of iron-bound diatomics on HydG is consistent with the recent FTIR spectroscopic studies [46]. A common thread amongst the majority of published work is that all three maturation proteins are essential, with HydE and HydG carrying out the ligand synthesis reactions and HydF serving as a scaffold or carrier.
Concluding remarks
The biosynthesis of the H-cluster of the [FeFe]-hydrogenase provides a fascinating example of how Nature synthesizes a complex organometallic cofactor in an aqueous environment from common metabolic precursors utilizing a small number of steps. Tyrosine is the substrate for HydG, and provides the CO and CN− ligands for the H-cluster via a complex radical-mediated reaction that remains the subject of active investigation. A key missing component in our understanding of [FeFe]-hydrogenase maturation is the identity of the precursor for the dithiomethylamine ligand of the H-cluster, and its mechanism of synthesis by HydE. A number of studies point to HydF serving as a scaffold or carrier of the 2Fe precursor to the H-cluster, although how the 2Fe precursor is assembled on HydF and delivered to HydA, and the role of GTP binding/hydrolysis in this process, remain a mystery.
References
Fontecilla-Camps JC, Amara P, Cavazza C, Nicolet Y, Volbeda A (2009) Nature 460:814–822
Mulder DW, Shepard EM, Meuser JE, Joshi N, King W, Posewitz C, Broderick B, Peters JW (2011) Structure 19:1038–1052
Vignais PM, Billoud B (2007) Chem Rev 107:4206–4272
Peters JW, Lanzilotta WN, Lemon BJ, Seefeldt LC (1998) Science 282:1853–1858
Nicolet Y, Piras C, Legrand P, Hatchikian CE, Fontecilla-Camps JC (1999) Structure 7:13–23
Silakov A, Wenk B, Reijerse E, Lubitz W (2009) Phys Chem Chem Phys 11:6592–6599
Erdem ÖF, Schwartz L, Stein M, Silakov A, Kaur-Ghumaan S, Huang P, Ott S, Reijerse EJ, Lubitz W (2011) Angew Chem Int Ed Engl 50:1439–1443
Berggren G, Adamska A, Lambertz C, Simmons TR, Esselborn J, Atta M, Gambarelli S, Mouesca JM, Reijerse E, Lubitz W, Happe T, Artero V, Fontecave M (2013) Nature 499:66–69
Esselborn J, Lambertz C, Adamska-Venkatesh A, Simmons T, Berggren G, Noth J, Siebel J, Hemschemeier A, Artero V, Reijerse E, Fontecave M, Lubitz W, Happe T (2013) Nat Chem Biol 9:607–609
Kubas GJ (2007) Chem Rev 107:4152–4205
Gordon JC, Kubas GJ (2010) Organometallics 29:4682–4701
Barton BE, Olsen MT, Rauchfuss TB (2008) J Am Chem Soc 130:16834–16835
Posewitz MC, King PW, Smolinski SL, Smith RD, Ginley AR, Ghirardi ML, Seibert M (2005) Biochem Soc Trans 33:102–104
Posewitz MC, King PW, Smolinski SL, Zhang L, Seibert M, Ghirardi ML (2004) J Biol Chem 279:25711–25720
Pierik AJ, Hulstein M, Hagen WR, Albracht SPJ (1998) Eur J Biochem 258:572–578
Tard C, Pickett C (2009) Chem Rev 109:2245–2274
Adamska A, Silakov A, Lambertz C, Rüdiger O, Happe T, Reijerse E, Lubitz W (2012) Angew Chem Int Ed 51:11458–11462
Mulder DW, Ratzloff MW, Shepard EM, Byer AS, Noone SM, Peters JW, Broderick JB, King PW (2013) J Am Chem Soc 135:6921–6929
King PW, Posewitz MC, Ghirardi ML, Seibert M (2006) J Bacteriol 188:2163–2172
McGlynn SE, Ruebush SS, Naumov A, Nagy LE, Dubini A, King PW, Broderick JB, Posewitz MC, Peters JW (2007) J Biol Inorg Chem 12:443–447
Boyer ME, Stapleton JA, Kuchenreuther JM, Wang C-W, Swartz JR (2007) Biotechnol Bioeng 99:59–67
Kuchenreuther JM, Stapleton JA, Swartz JR (2009) PLoS One 4:e7565
Mulder DW, Ortillo DO, Gardenghi DJ, Naumov A, Ruebush SS, Szilagyi RK, Huynh BH, Broderick JB, Peters JW (2009) Biochemistry 48:6240–6248
Mulder DW, Boyd ES, Sarma R, Lange RK, Endrizzi JA, Broderick JB, Peters JW (2010) Nature 465:248–251
Kuchenreuther JM, Guo Y, Wang H, Myers WK, George SJ, Boyke CA, Yoda Y, Alp EE, Zhao J, Britt RD, Swartz JR, Cramer SP (2013) Biochemistry 52:818–826
Brazzolotto X, Rubach JK, Gaillard J, Gambarelli S, Atta M, Fontecave M (2006) J Biol Chem 281:769–774
McGlynn SE, Shepard EM, Winslow MA, Naumov AV, Duschene KS, Posewitz MC, Broderick WE, Broderick JB, Peters JW (2008) FEBS Lett 582:2183–2187
Shepard EM, McGlynn SE, Bueling AL, Grady-Smith C, George SJ, Winslow MA, Cramer SP, Peters JW, Broderick JB (2010) Proc Natl Acad Sci USA 107:10448–10453
Kuchenreuther JM, Britt RD, Swartz JR (2012) PLoS One 7:e45850
Berto P, De Valentin M, Cendron L, Vallese F, Albertini M, Salvadori E, Giacometti GM, Carbonera D, Costantini P (2012) Biochim Biophys Acta 1817:2149–2157
Czech I, Silakov A, Lubitz W, Happe T (2010) FEBS Lett 584:638–642
Czech I, Stripp S, Sanganas O, Leidel N, Happe T, Haumann M (2011) FEBS Lett 585:225–230
Vallese F, Berto P, Ruzzene M, Cendron L, Sarno S, De Rosa E, Giacometti GM, Costantini P (2012) J Biol Chem 287(43):36544–36555
Cendron L, Berto P, D’Adamo S, Vallese F, Govoni C, Posewitz MC, Giacometti GM, Costantini P, Zanotti G (2011) J Biol Chem 286:43944–43950
Joshi N, Shepard EM, Byer AS, Swanson KD, Broderick JB, Peters JW (2012) FEBS Lett 586:3939–3943
Pilet E, Nicolet Y, Mathevon C, Douki T, Fontecilla-Camps JC, Fontecave M (2009) FEBS Lett 583:506–511
Driesener RC, Challand MR, McGlynn SE, Shepard EM, Boyd ES, Broderick JB, Peters JW, Roach PL (2010) Angew Chem Int Ed Engl 49:1687–1690
Shepard EM, Duffus BR, McGlynn SE, Challand MR, Swanson KD, Roach PL, Peters JW, Broderick JB (2010) J Am Chem Soc 132:9247–9249
Kuchenreuther JM, George SJ, Grady-Smith CS, Cramer SP, Swartz JR (2011) PLoS One 6:e20346
Driesener RC, Duffus BR, Shepard EM, Bruzas IR, Duschene KS, Coleman NJ-R, Marrison APG, Salvadori E, Kay CWM, Peters JW, Broderick JB, Roach PL (2013) Biochemistry 52:8696–8707
Walsby CJ, Ortillo D, Broderick WE, Broderick JB, Hoffman BM (2002) J Am Chem Soc 124:11270–11271
Walsby CJ, Hong W, Broderick WE, Cheek J, Ortillo D, Broderick JB, Hoffman BM (2002) J Am Chem Soc 124:3143–3151
Walsby CJ, Ortillo D, Yang J, Nnyepi M, Broderick WE, Hoffman BM, Broderick JB (2005) Inorg Chem 44:727–741
Tron C, Cherrier MV, Amara P, Lydie M, Fauth F, Fraga E, Correard M, Fontecave M, Nicolet Y, Fontecilla-Camps JC (2011) Eur J Inorg Chem 2011:1121–1127
Kuchenreuther JM, Myers WK, Stich TA, George SJ, Nejatyjahromy Y, Swartz JR, Britt RD (2013) Science 342:472–475
Kuchenreuther JM, Myers WK, Suess DLM, Stich TA, Pelmenschikov V, Shiigi SA, Cramer SP, Swartz JR, Britt RD, George SJ (2014) Science 343:424–427
Rubach JK, Brazzolotto X, Gaillard J, Fontecave M (2005) FEBS Lett 579:5055–5060
Nicolet Y, Rubach JK, Posewitz MC, Amara P, Mathevon C, Atta M, Fontecave M, Fontecilla-Camps JC (2008) J Biol Chem 283:18861–18872
Nicolet Y, Amara P, Mouesca J-M, Fontecilla-Camps JC (2009) Proc Natl Acad Sci USA 106:14867–14871
Duffus BR, Hamilton TL, Shepard EM, Boyd ES, Peters JW, Broderick JB (2012) Biochim Biophys Acta 1824:1254–1263
Shepard EM, Boyd ES, Broderick JB, Peters JW (2011) Curr Opin Chem Biol 15:319–327
Acknowledgments
Work on [FeFe]-hydrogenase and its maturation in the Broderick and Peters laboratories is supported by the U.S. Department of Energy (DE-FG02-10ER16194 to J.B.B., J.W.P., and E.M.S.) and U.S. Air Force Office of Scientific Research (FA9550-11-1-2-0218 to J.W.P.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Broderick, J.B., Byer, A.S., Duschene, K.S. et al. H-Cluster assembly during maturation of the [FeFe]-hydrogenase. J Biol Inorg Chem 19, 747–757 (2014). https://doi.org/10.1007/s00775-014-1168-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-014-1168-8