
Abstract
Nitrophorin 2 (NP2), one of the four NO-storing and NO-releasing proteins found in the saliva of the blood-sucking bug Rhodnius prolixus, has a more ruffled heme and a high preference for a particular heme orientation (B) compared with nitrophorin 1 and nitrophorin 4, which show not a preference (A to B ratio of approximately 1:1), suggesting that it fits more tightly in the β-barrel protein. In this work we have prepared a series of “belt” mutants of NP2(D1A) and (ΔM0)NP2 aimed at reducing the size of aromatic or other residues that surround the heme, and investigated them as the high-spin aqua and low-spin N-methylimidazole complexes. The belt mutants included Y38A, Y38F, F42A, F66A, Y85A, Y85F, Y104A, I120T, and a triple mutant of NP2(D1A), the F42L, L106F, I120T mutant. Although I120 has been mainly considered to be a distal pocket residue, CδH3 of I120 lies directly above the heme 3-methyl, at 2.67 Å, of heme orientation B, or the 2-vinyl of A, and it thus plays a role as a belt mutant, a role that turns out to be extremely important in creating the strong favoring of the B heme orientation [A to B ratio of 1:14 for NP2(D1A) or 1:12 for (ΔM0)NP2]. The results show that the 1D 1H NMR spectra of the high-spin forms are quite sensitive to changes in the shape of the heme binding cavity. The single mutation I120T eliminates the favorability of the B heme orientation by producing a heme A to B orientation ratio of 1:1, whereas the single mutation F42A reverses the heme orientation from an A to B ratio of 1:14 seen for NP2(D1A) to 10:1 for NP2(D1A,F42A). The most extreme ratio was found for the triple mutant of NP2(D1A), NP2(D1A,F42L,L105F,I120T), in which the A to B ratio is approximately 25:1, a ΔG change of about −3.5 kcal/mol or −14.1 kJ/mol with respect to NP2(D1A). The seating of the heme is modified as well in that mutant and in several others, by rotations of the heme by up to 4° from the seating observed in NP2(D1A), in order to relieve steric interactions between a vinyl β-carbon and a protein side chain, or to fill a cavity created by replacing a large protein side chain by a much smaller one; the latter was observed for all tyrosine to alanine mutants. These relatively small changes in seating have a measurable effect on the NMR spectra of the mutants, but are indeed minor in terms of overall seating and reactivity of the NP2(D1A) protein. The 1H NMR resonances of the hemin substituents of the low-spin N-methylimidazole complexes of NP2(D1A,F42L,L105F,I120T) as well as NP2(D1A,I120T), NP2(D1A,Y104A), and NP2(D1A,F42A) have been assigned using natural abundance 1H{13C} heteronuclear multiple quantum correlation and 1H–1H nuclear Overhauser effect spectroscopy spectra.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The nitrophorins are a group of nitric oxide (NO)-storing and NO-carrying heme proteins found in the saliva of some species of blood-sucking insects [1]. One of these insects is Rhodnius prolixus (the “kissing bug”), the adult insect of which has four salivary nitrophorins [nitrophorin 1 (NP1), nitrophorin 2 (NP2), nitrophorin 3 (NP3), and nitrophorin 4 (NP4) in order of decreasing abundance] that store NO via a ferriheme–nitrosyl complex and keep it stable for long periods of time [2, 3]. The NO is produced by a constitutive nitric oxide synthase enzyme which is present in the epithelial cells of the salivary glands [4]. When injected into the tissues of the bug’s victim, a combination of approximately 100-fold dilution and pH rise (from a pH of approximately 5–6 in the salivary glands to approximately 7.3–7.4 in the tissues) causes dissociation of NO. The NO diffuses through the tissues to the nearby capillaries and causes vasodilation (and inhibition of platelet aggregation), thereby allowing more blood to be transported to the site of the wound [5, 6]. In addition, the ferriheme centers of the Rhodnius nitrophorins are able to bind histamine, which is released by mast cells and platelets of the victim in response to the wound. This histamine would otherwise cause swelling, itching, and the beginning of the immune response of the victim; its binding to the nitrophorins hence prevents the detection of the insect for a period of time [7]. These properties of the nitrophorins of the insect assist the bug in obtaining a sufficient blood meal in a minimum amount of time. Of the four Rhodnius nitrophorins of the adult insect, NP2 is unique, for in addition to its NO-releasing and histamine-binding roles, it also has anticoagulant activity, via heme-independent inhibition of the factor Xase complex [8–10].
The structures of Rhodnius nitrophorins bound to various ligating species have been determined by X-ray crystallography for NP1 [11–13], NP2 [14–16], and NP4 [17–25]. These structures show the heme to be located inside a β-barrel, with the propionate groups protruding into the aqueous medium. This structure is unique for heme proteins, which more commonly have α-helical globin or four-helix bundle folds [26, 27]. The ferriheme prosthetic group is bound to the protein via a histidine ligand, and the sixth coordination site is available to bind NO, histamine, or other ligating species, and if no other ligating species is added, water, to yield a high-spin Fe(III) complex. Ligand binding has been investigated by a number of spectroscopic techniques [12, 28–46], including infrared [12, 28], resonance Raman [28], EPR [30], NMR [12, 31–41], and Mössbauer [41, 43] spectroscopies, nuclear inelastic scattering [43], stopped-flow [44] and laser flash [45, 46] kinetics, and spectroelectrochemistry [12, 25, 32, 35, 44]. The nitrophorins store and transport NO via a ferriheme–nitrosyl complex, which is bound to NO with stabilities that facilitate release on dilution in biological tissues (K d ~ 5–900 nM) [25]. Typically, other heme proteins have much more positive reduction potentials than the nitrophorins, and these ferriheme–nitrosyl complexes are unstable with respect to reduction to Fe(II)–NO in the presence of excess NO [47], and bind NO irreversibly (K d in the picomolar to femtomolar range [45]).
The nitrophorins all have two conserved leucine residues in the distal pocket where NO, histamine, and other ligating species can bind, which have their side chains positioned above and in contact with the heme near the iron binding site, as shown in Fig. 1, where they may possibly contribute to the extensive distortion of the heme from planarity which is particularly extreme in NP2, but is also present in NP1 and NP4. Unfortunately, it has not been possible to obtain crystal structures of NP3 thus far, but because of the similarity in the protein sequences and the proton NMR shifts of protein side chains of NP2 and NP3 [39], we surmise that NP3 has a similarly nonplanar heme, as well as a heme binding pocket extremely similar to that of NP2. The nonplanarity is believed to affect the FeIII/FeII midpoint potential, and thus the reactivity of the ferriheme center toward NO [32]. The Leu122(123) side chain is situated near the opening to the ligand binding pocket, whereas the Leu132(133) side chain is buried deep inside the pocket, and is believed to be largely responsible for the heme nonplanarity and midpoint potential [19, 32].

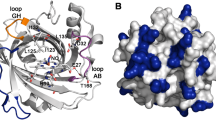
The heme pockets of a D1A mutant of nitrophorin 2 (NP2) complexed with NH3, Protein Data Bank (PDB) file 2EU7 [15] (left), and nitrophorin 4 (NP4) complexed with NH3, PDB file 1X8P [22] (right), with the residues around the heme, four of which differ between the two proteins, highlighted in CPK mode [L106 vs F107 (yellow), I120 vs T121 (apple green), S40 vs A42 (brown), and F42 vs L44 (orange)]. Also shown are the “belt” aromatic residues [Y104(105) (royal blue), Y85(F87) (pink), E53(55) (black), Y38(40) (grass green), L122(123) (aqua), and L132(133) (olive)], all in ball and stick mode
In addition to the two conserved leucine residues in the NP2 and NP3 distal pocket, there is a third residue which also is positioned above and in contact with the heme near the distal ligand binding site, also deep inside the distal pocket. This residue is an isoleucine (I120) for NP2 and NP3 (Fig. 1), but a threonine (T121) for NP1 and NP4. The crystal structure of NP2(D1A)-NH3 (PDB file 2EU7 [15]) shows that the I120 Cδ1 methyl carbon is only 3.50 Å from the heme 3-methyl carbon, meaning that their methyl protons are slightly less than 2.59 Å apart, or essentially within van der Waals contact. In contrast, for the B heme orientation of NP4, the Cγ2 methyl carbon of T121 is 4.66 Å from the 3-methyl carbon of the B heme orientation, and their protons are 3.32 Å apart (PDB file 1KOI [19]), or 4.36 Å from the 1-methyl carbon of the A heme orientation, and their protons are 3.09 Å apart. This means that T121 cannot contribute to the ruffling of the heme of NP4, and indeed, X-ray crystal structures show the heme of NP4 [17–22] is less ruffled than that of NP2 [14, 15] (Table S1). Because of the large difference in the sizes of the side chains of isoleucine and threonine, as well as the difference in their hydrophobicity, we wondered what the role of this residue is in determining the properties of the nitrophorins, in particular the heme seating and the ratio of heme orientations, A and B, which exist because of the unsymmetrical placement of the two vinyl groups of protohemin IX (Fig. 2). In the wild-type nitrophorins, NP1 and NP4 show no preference for A or B (A to B ratio of approximately 1:1), whereas NP2 has an A to B ratio of 1:25 at equilibrium and NP3 has a ratio of about 1:4. Therefore, we recently studied the effects of the size of the I120/T121 residue in detail [38] by preparing the I120T and I120V mutants of wild-type NP2 and the T121S, T121L, and T121I mutants of wild-type NP1 and found that the two large side chains produce an equilibrium ratio of A and B isomers which favors the B orientation, 2:3 and 1:2, respectively. However, since the presence of M0 in wild-type NP2 [hereafter called (M0)NP2] and wild-type NP1 [hereafter called (M0)NP1] prevents the A–B and G–H loops from closing [35], we have also investigated the I120T mutant of NP2(D1A) and native N-terminus NP2 [hereafter called (ΔM0)NP2], as well as a number of other site-directed mutants, in this work.

Heme orientations A and B, with H57 behind the heme, and I120 (orange), L122 (yellow), and L132 (yellow) in front of the heme. Dotted lines β,δ meso line, solid lines histidine imidazole plane orientation
(M0)NP1 and NP4 both have nearly equal ratios of heme orientations A and B, whereas (M0)NP2 and (M0)NP3 have been reported to have A to B ratios of 1:8 and 1:4 at equilibrium for the high-spin forms, respectively [33]. However, in recent work we have found that with equilibration times of 8 days or more, the A to B ratio of fully equilibrated (M0)NP2 is actually 1:25 [40], whereas that of NP2(D1A) is 1:14 [35] and that of (ΔM0)NP2 is similar, at 1:12 [39]. Thus, for high-resolution, multidimensional NMR studies, NP2 is by far the most favorable Rhodnius nitrophorin to investigate, because it has a large difference in the abundance of the two heme isomers [31–41]. For example, at pH 7.0–7.5 with an A to B ratio of 1:14, the resonances of the 7.1 % A orientation of NP2(D1A) do not interfere with assignment of the protein resonances of the B-orientation heme-containing protein in the multidimensional (2D to 4D) 1H,13C,15N NMR experiments, or the use of (ΔM0)NP2 for conducting dynamics measurements by NMR spectroscopy [48]. For the other three Rhodnius nitrophorins it would be necessary to use the “symmetrical hemin” (2,4-dimethyldeuterohemin) in order to assign the protein resonances. And as will become clear throughout this article, the two extra carbons of the two vinyl groups actually affect a number of the thermodynamic and spectroscopic properties of the nitrophorins.
In addition to the three distal pocket side chains of all four nitrophorins of the adult insect, discussed above, a number of residues are positioned like a belt around the inside of the β-barrel, as shown in Fig. 1, in such a way that they may interact with the heme and affect the heme A to B orientation ratio and the tightness of the heme fit inside the β-barrel, and thus the degree of ruffling of the heme. Among these, there are several sequence differences between NP1 and, NP4 on one hand and NP2 and NP3 on the other, for which the side chains are different in size, for example, A42 as compared with S40, L44 as compared with F42, F107 as compared with L106, and I120 as compared with T121, respectively. We have already investigated the S40A mutant of (M0)NP2 and the A42S mutant of (M0)NP1 and found that neither mutation changed the A to B ratio or the seating of the heme, and thus this position is not important in controlling the heme orientation or seating [38]. However, we have also investigated the L44F mutant of (M0)NP1 and found that it has significant chemical exchange processes in the high-spin state that affect the appearance of the 1D NMR spectra of this mutant [36, 38], as well as in the low-spin imidazole complex [38]. In this work we have continued the investigation of the differences in residues between the two groups of nitrophorins, shown in space-filling mode in Fig. 1. Many other “belt” residues are conserved, or nearly so, among the nitrophorins, including V24(25), F27(Y28),Footnote 1 V34(36), E53(55), L55(57), Y85(F86), and Y104(105), and we have probed some of these in this study as well. Finally, the presence of M0 in the case of (M0)NP1, (M0)NP2, and (M0)NP3, as compared with NP4, which has A1 as its native N-terminal residue, also provides a major difference in the conformation of the A–B and G–H loops of the four major nitrophorins [35], and possibly also the size and shape of the heme binding pocket. Thus, we have investigated this major difference further within the series of proteins presented herein, by preparing a number of belt mutants in the D1A construct of NP2, which lacks M0 [35], and in the (ΔM0)NP2 construct, which also lacks M0 and which has its native D1 as the first residue [40]. In this work we report the 1H NMR spectra of the belt mutants of high-spin and low-spin NP2(D1A) and (ΔM0)NP2.
Materials and methods
Protein sample preparation
The cloning and expression plasmids used for the D1A mutants were those reported previously [14, 35]. Additional mutant genes [for the NP2(D1A,Belt series), where Belt is Y38A, F42A, F66A, Y85A, Y104A, and I120T, as well as the triple mutant of NP2(D1A), F42L,L106F,I120T] were prepared according to standard genetic engineering methods, and expressed as reported previously [14, 35]. Several additional mutants, where Belt is Y104F, Y104H, and Y104W, would not fold properly, either in the (M0) or in the (D1A) construct, under the many folding conditions tried. Attempts to express and fold NP4(Y105F) also failed. The corresponding (ΔM0)NP2 mutants Y38F, Y85F, Y104A, and I120T were prepared by methods reported recently [40], in combination with the usual site-directed mutagenesis techniques [14, 35]. The (ΔM0)NP2(Y104F) mutant gene was also constructed and expressed, but the protein could not be folded. The Escherichia coli expressed mutant proteins were purified as described previously [14, 35, 40], and were stored in lyophilized form at −80 °C until use. The (ΔM0)NP2 mutant proteins, which were bound to protohemin that was produced during expression, were preliminarily characterized by UV–visible spectroscopy, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry, and 1D 1H NMR spectroscopy. The preliminary 1D NMR spectra showed that all (ΔM0)NP2 mutants contained a small amount of some adventitious ligand that was produced during expression and/or purification, and when these proteins were expressed in small quantity (4–6-L growths), it was found to be impossible to remove this ligand by simple purification techniques used previously [14, 35], which produced variable amounts of low-spin heme protein signals in the high-spin samples.
NMR data collection
NMR samples were prepared in D2O containing 50 mM phosphate buffer at pH* 7.0 (pH* indicates that the pH was read using a standard pH meter and was not corrected for the deuterium isotope effect). NMR spectra were collected over the temperature range 25–35 °C, and occasionally at a temperature as high as 37 °C with the proton chemical shifts referenced to residual water or to 2,2-dimethyl-2-silapentane-5-sulfonate sodium salt [49]. Natural abundance 1H{13C}heteronuclear multiple quantum correlation (HMQC) experiments were recorded at a proton Larmor frequency of 600.1299902 MHz and 30 °C with a Bruker AVANCE spectrometer, using a 5-mm inverse-detection probe with decoupling during acquisition. A relaxation delay of 0.2 s and a refocusing time of 0.0032 s (J = 200 Hz) were used. For the water-elimination Fourier transform (WEFT) nuclear Overhauser effect spectroscopy (NOESY) experiments, recorded at 600.1299902 MHz and 30 °C with the same spectrometer, a relaxation delay of 0.70 s and a recovery delay of 0.125 s were used. The mixing time for the NOESY experiments was 0.012 s. All 2D spectra were collected with 1,918 or 3,072 data points in t 2 for WEFT-NOESY and HMQC experiments, respectively, and with 512–540 blocks in t 1 with 80–512 scans per block.
Results and discussion
Use of “angle plots”
Before discussing the 1H NMR spectra of the mutants of this study, we introduce the “angle plots,” which are useful tools for analyzing the NMR spectra of ferriheme proteins having at least one histidine or exogenous imidazole bound to iron(III). The angle plots are based on the effect of spin delocalization from unpaired electrons in the d xz and/or d yz (d π ) orbitals of paramagnetic Fe(III) to the porphyrin ring by L → Fe π donation, in combination with P → Fe π donation to the same d π orbital, where L represents the histidine imidazole nitrogen π orbital nodal plane, and P represents the porphyrin 3e(π) orbital of proper symmetry to interact with both the particular d π orbital and the L π orbital [31, 50]. These angle plots were developed first for low-spin ferriheme proteins [50], and were later extended to high-spin ferrihemes [31]. (They have also been developed for methionine-ligated ferrihemes [51, 52], since the orientation of the π-symmetry p-orbital nodal plane of the sulfur plays the same role for cytochromes c as does the π-symmetry p-orbital nodal plane of the binding nitrogen of the imidazole ring of histidine ligands. When both histidine and methionine are present, it is the methionine π-symmetry nodal plane orbital which dominates the pattern of spin delocalization.) The idea behind the angle plots is that one can write two possible limiting structures of the 3e(π) molecular orbitals of the porphyrin ring, one set of which has a nodal plane passing through opposite nitrogen atoms, and the other set of which has a nodal plane passing through opposite meso carbons, and is thus oriented at 45° at the first set mentioned [50–54]. From these two, one can realize that it should also be possible for the nodal plane to take on the orientation of the other two porphyrin nitrogens, or the other two meso positions, thus covering a total of 180°, and in fact, any arbitrary orientation within that 180°. From that point it is only a small leap of understanding to realize that a p orbital on a ligand nitrogen which is part of an aromatic ring (like that of the nitrogen of the imidazole ring of histidine which binds to iron) can control the delocalization of spin density from the d xz or d yz orbital of Fe(III) to specific points on the periphery of the porphyrin ring, which will thus have more or less spin density delocalized to each position, depending on the particular angle of that nodal plane. Thus, our original hypothesis can be said to be that the nodal plane of a π-donor axial ligand binding to a heme defines the orientation of the e-symmetry d π orbital of iron and the nodal plane of the 3e(π) orbital of the porphyrin ring determines the pattern of spin density delocalization to heme substituents. For low-spin ferrihemes, the electron spin of the half-filled d π orbital can thus be delocalized efficiently to the porphyrin ring [50], whereas for high-spin ferrihemes, with five unpaired electrons, one of the d π orbitals will interact with the axial ligand, and the other interacts with the porphyrin ring, thus introducing a 90° shift into the angle plot of high-spin ferrihemes as compared with low-spin ferrihemes [31, 55]. The angle plots are useful for answering questions about heme seating, since they relate the order and relative spacings of the methyl resonances of protohemin-containing proteins of either S = 5/2 spin state (Fig. 3), or S = 1/2 spin state (Fig. S1) to the angle of the imidazole nodal plane of the coordinated histidine [55] (or a planar exogenous ligand used to produce a low-spin complex).

“Angle plot” for high-spin ferrihemes [31, 55], adjusted to the chemical shift scale of the high-spin NP2(D1A) complex, which has 5M and 8M at the same chemical shift. This is the best average angle plot that can be constructed for the NP2(D1A) mutants of this study, although the chemical shift scale differs somewhat for each mutant. The polarity of the heme pocket differs for each mutant, likely because of different numbers of water molecules being present in place of the protein side chain originally present in NP2(D1A) or (ΔM0)NP2. Lines 1, 3, 5, and 8 represent the chemical shifts of the four methyl groups of protohemin, as a function of the angle of the axial histidine imidazole plane from the x-axis of the heme in A or B orientation shown in Fig. 2, projected on the heme, as viewed from above. The angles of the axial histidine imidazole plane shown in Fig. 2 are projected onto the angle plot at 125° and 145°, and a third line is provided at 135°, the crystallographic angle for NP2(D1A), PDB file 2EU7 [15]. The average of the four lines is also plotted (dashed line)
A remarkable range of ferriheme proteins has been found to obey the angle plots [50–60]. In these angle plots, we have not made corrections for the presence of aromatic residues near the heme methyls, because for mainly α-helical heme proteins (myoglobins, cytochromes b 5) there are very few aromatic residues near the hemes, and all of those which are present are phenylalanines; when present, they tend to be at oblique angles to the heme methyls, and thus are not properly positioned to contribute to a ring current shift (see below). Peroxidases, which tend to have two phenylalanines (and no tyrosines or tryptophans), and cytochromes c, which tend to have a number of tyrosines and tryptophans as well as phenylalanines, are positioned so as to cause weak ring current shifts for 5M and 8M (F172 and 179, respectively, for horseradish peroxidase, PDB file 1ATJ), whereas the two near 1M (F142, F143) are too far away to have much effect, and the two near 3M (F45, F152) are at quite oblique angles. For cytochromes c, W59 and Y67 are near 8M, Y46 is near 5M, and F10 is near 1M (tuna, PDB file 3CYT), and for horse heart cytochrome c (PDB file 1HRC), the same if found, except that F46 is at a very oblique angle and probably does not contribute significantly. In contrast, for the β-barrel-structured nitrophorins, there is one or more aromatic residue near each of the heme methyls [F27 near 1M, F42 near 3M, Y85, Y104, and F66 near 5M, and Y38 and F66 near 8M (Fig. 1), and similarly for NP1 and NP4, except for changes in the residue numbers and F27 and Y85 of NP2 becoming Y28 and F86 for NP1 and NP4] for both heme orientation B and, with slight rearrangements of the angles of F27 and F42, for heme orientation A.
Case [61], in an article which also develops density functional theory methods for methane molecules placed in a variety of positions near aromatic rings of the type found in proteins and nucleic acids, and much earlier Haigh and Maillon [62] have reviewed the empirical equations developed from Hückel molecular orbital theory for calculating ring currents near specific sites in proteins. We have used the Hückel approach to evaluate the ring current shifts resulting from the aromatic residues near each of the four heme methyls of NP2 in the B heme orientation. The general form expected for ring current contributions to chemical shifts from the Hückel point of view is
where r is the vector from the observed proton to the aromatic ring, G(r) is a geometric factor and i and B are constants. The geometric factor is
where r i and r j are the distances from ring atoms i and j to the proton and s ij is the area of the triangle formed by atoms i and j and the proton point projected onto the plane of the aromatic ring. The sum is over the bonds in the ring. For a methyl group, which spins very rapidly at solution NMR temperatures, all three protons must be treated.
Groups which are nearly perpendicular to the proton or group of protons do not contribute, although they would contribute to deshielding in the Johnson–Bovey treatment of ring currents [63] found in many organic chemistry textbooks. Thus, in the case of NP2, Y104 and F66 would not be included because the protons of interest (heme methyl protons) lie far from the perimeter of the ring of the aromatic residue, and are perpendicular to the heme methyl. Thus, we have calculated the values of σ rc for F27 near 1M, F42 near 3M, Y85 near 5M, and Y38 near 8M, and found that σ rc for 1M is −0.66 ppm, for 3M is −0.54 ppm, for 5M is −0.52 ppm, and for 8M is −0.46 ppm, or as a summary, the ring current contributions to the nitrophorins are σ rc ~ −0.5 ± 0.1 ppm for all four methyls, which is certainly within any experimental error that we might realistically assign to the chemical shifts obtained from the angle plot. Therefore, for the nitrophorins, although there appear to be ring currents, they act as a constant contribution that simply shifts the numerical parts per million scale. Thus, contributions of aromatic ring currents from phenylalanines, tyrosines, and tryptophans are not considered in the angle plots. It has never been claimed that the angle plots predict the chemical shifts to high accuracy; that is the reason that the chemical shift scale was removed soon after publication of the original article [50, 55]. However, with regard to the angles where heme methyl resonances cross each other, we should expect that in the vast majority of cases, those crossing points should be useful indicators of whether the seating of the heme remains the same or changes with a particular mutation, because they arise from the angular relationship of the heme methyls to each other and the orientation to the nodal plane of the ligand.
If one of those aromatic residues of NP2(D1A) or (ΔM0)NP2 is mutated to alanine, two things will happen simultaneously: The ring current shift of the aromatic residue will be removed, and a number of water molecules will undoubtedly enter to fill the hole created by removal of the large side chain. Those water molecules are expected to change the polarity of the region where they are inserted.
In the case of high-spin ferriheme proteins, since the histidine ligand is provided by the protein, the histidine is likely not able to change its orientation significantly and is likely still able to bind to the metal, because it is locked into a particular rotamer and cannot rotate significantly about its Nε–Fe bond. In these cases, it will likely be the heme ring which rotates, thus changing its “seating” in order to fit into the mutated heme pocket shape, rather than the histidine imidazole. Some rotations may be by a relatively small number of degrees, as is the usual case for the nitrophorins (as marked on the two angle plots), whereas others can be very large (approximately 110°), as for one of the heme seatings of Pseudomonis aeruginosa heme oxygenase bound to its substrate (heme) [56].
For the nitrophorins, X-ray crystallography has shown that H57 (H59 for NP1 and NP4) is oriented such that its imidazole plane lies within ±1° of the β,δ meso line [11–23] or 135 ± 1° from the x-axis of Fig. 2, measured in a counterclockwise direction for heme orientation A and in a clockwise direction for heme orientation B. Note that φ = 135° is the precise angle where the chemical shifts of 8M and 5M are calculated to cross each other (Fig. 3 for high spin, Fig. S1 for low spin). Thus, if 8M and 5M of a high-spin ferriheme complex have the same chemical shift, we know that the angle is precisely 135°. For example, for NP2(D1A), 5M and 8M occur together at 64.3 ppm. X-ray crystallography of NP2(D1A)-NH3 (a low-spin complex, but it still tells us the orientation of H57) shows that the H57 ligand plane lies exactly over the meso β–meso δ line, in the low-spin case for the exogenous ligands NH3 and CN−, which contribute no ligand plane to the complex. For N-methylimidazole (NMeIm) as a ligand, however, we have found during our studies of the nitrophorins that this exogenous ligand (or any other exogenous imidazole, including imidazole, 5-methylimidazole, and 2-methylimidazole), rather than H57(59) or an average of the two, dominates the NMR chemical shifts of the heme methyls, and NMeIm usually sits at φ ~ 150–165° for B or φ ~ 105–120° for A, depending on the mutation. For the “parent,” NP2(D1A)-NMeIm or (ΔM0)NP2-NMeIm, the angle is 161–162° for B and 109–110° for A, on the basis of the relative spacings worked out for the quantitative scale for B (Fig. 3). The predicted shifts for A are quite similar to the observed shifts, although they are not identical. This is, in the opinion of the authors, why it is not worthwhile trying to construct exact scales for a given protein, as we have in this particular case at the insistence of a reviewer, because they will never fit another protein exactly. Although the low-spin complexes do not confirm for us what the orientation of the H57 imidazole plane is, they provide unambiguous assignments for most or all of the heme substituents, which can readily be transferred back to the high-spin complexes, as discussed below.
If the chemical shifts of 8M and 5M are not the same, then the heme methyl resonances can be assigned by “walking around the heme” of the low-spin complexes, using NOESY cross peaks between adjacent heme substituents, and natural abundance 1H{13C} HMQC spectra, which clearly identify heme methyls [64] (as well as propionate Hα and Hβ, but less so vinyl Hα and Hβ, which are often not well resolved). These HMQC assignments are particularly important for the substituents which are buried in the diamagnetic protein envelope, and it is easy to tell a methyl from a methylene. This can be done only for the low-spin complexes because of the rapid electron spin relaxation of the high-spin complexes, and thus it is typically necessary to add NMeIm, or some other not too strongly binding species, so that the low-spin methyl assignments can be made and then can be transferred to the high-spin spectra by 1D saturation transfer difference (STD) spectroscopy. [The STD experiments require a separate sample, which has 0.25 equiv or less of NMeIm present, so as to be able to observe both high-spin (broad) and low-spin (much sharper) resonances.] The assignments can then be used to determine the angle of the histidine imidazole nodal plane with respect to the heme axis system. Since the changes in angle are relatively small (1–4°), for high-spin samples (Fig. 3) heme orientational isomer A always has a larger methyl resonance spread than heme orientational isomer B when both heme orientations are present to an abundance where the methyls of both can be assigned. The A heme orientation usually has the order 5M > 8M > 1M > 3M or 5M = 8M > 1M > 3M, whereas the B heme orientation usually has the order 8M > 5M > 1M > 3M or 8M = 5M > 1M > 3M. (Note that these orders are specifically for the nitrophorins, and may be entirely different for other heme proteins!)
The angle plots for 5M and 1M for both high-spin and low-spin ferrihemes have the same phase (they are 180° apart on the heme ring), yet the plot for 1M is offset, thus always predicting that 1M will have a smaller chemical shift than 5M. Less obvious, because of the difference in phase of the 3M and 8M lines, is the fact that the 3M plot is also offset to lower chemical shift from the 8M plot. These offsets were introduced when the angle plots were created, because of the substituent effects of the vinyls, as compared with the alkyl side chains of the propionates [55], since protohemin biscyanide has methyl chemical shifts in the order 8M ~ 5M > 3M ≳ 1M (16.3, 15.9, 12.3, and 10.3 ppm) [65]; thus, offsets of some amount between 5M and 1M, and the same amount (although it does not have to be the same) between 8M and 3M at 0° and 180° were introduced into the angle plots [50]. Potential changes in this separation, especially for 5M and 1M, were discussed (especially in regard to cytochrome c hemes, where the vinyls have become ethyl groups, with the substituent effect having decreased to nearly zero), and were shown in the first “angle plot” article [55]. The numerical chemical shift scale was later removed to increase generality [55], but we have continued to think of 5M and 1M as having a significant difference in chemical shift at any angle of the axial ligand plane. At times we have found that this was experimentally not the case, even with 1M > 5M for the B heme orientation of a few specific mutants [38]. For both high-spin and low-spin mutants presented in this article, it is clear that the 5M–1M chemical shift difference actually varies by quite a bit, as seen in Tables 1, 2, and 3, 1.7–9.2 ppm for high-spin nitrophorins and 0.97–2.3 ppm for low-spin nitrophorins. This variation may be due to changes in the degree of ruffling of the heme, or to changes in the degree of conjugation of the vinyl groups with the heme, or to water molecules changing the polarity near one methyl and not another, or to other factors which have not as yet been identified. Further study of additional mutants will be required in order to understand the reason(s) for the variation. Until that time, we will continue to use the angle plots shown in Fig. 3 or Fig. S1.
NMR characterization of the high-spin complexes of the NP2(D1A,Belt) and (ΔM0)NP2(Belt) mutants
We have shown, by preparation of the D1A mutant of NP2, for which the E. coli methionine aminopeptidase is able to cleave Met0 [35] as it does for NP4 (whose native first residue is alanine), that the large Met0 side chain interferes with closure of the A–B loop. This A–B loop, in the absence of Met0, has been shown to take up additional conformations [48], which, along with the G–H loop [17–22], have been suggested to reduce the rates of NO and histamine release [44, 45] and of heme A to B orientational equilibration at low pH [35]. The D1A mutant of NP2 has a significant effect on heme orientation (A to B ratio of approximately 1:14 [35] as compared with 1:25 for (M0)NP2 [40]) and axial ligand complex stability [35, 40], but a very small effect on heme seating [39], as reported previously [35]. More recently we prepared (ΔM0)NP2, and found that its A to B heme orientation ratio is similar, 1:12 [40]. One of our first findings about the two N-terminus proteins was that the NP2(D1A) and (ΔM0)NP2 1D 1H NMR spectra are essentially identical in the high-spin (S = 5/2) state (Fig. 4) and also in the low-spin (S = 1/2) state [40], thus making it possible to compare NMR spectra of mutants of NP2 in the two constructs directly.

1H NMR spectra of high-spin NP2(D1A), (ΔM0)NP2, NP2(D1A,Belt), where Belt represents F42A, Y104A, Y85A, Y38A, or (ΔM0)NP2(Belt), where Belt represents Y104A, Y85F, or Y38F, as well as the distal pocket mutant I120T with both N-termini (A1 and D1) and the proximal pocket mutant F66A with the D1A N-terminus, all recorded at 600 MHz, pH* 7.0, and 25 °C, showing large effects resulting from mutation of Y38, Y104, or Y85 to alanine (but much smaller changes, and a somewhat smaller change in heme seating on mutation of F66 to alanine, but a very large change in the heme orientation ratio on mutation of I120 to threonine (A to B ratio of approximately 1:1), mutation of F42 to alanine (A to B ratio of approximately 10:1), mutation of Y104 to alanine (A to B ratio of approximately 1:10), and mutation of Y85 to alanine (A to B ratio of approximately14:1). For both the I120T mutant and the F42A mutant, the spectra are sharp, suggesting that there is little or no dynamics involved in heme seating. For the Y104A and Y85A mutants, some of the propionate resonances are very broad, suggesting that rotations about the β-pyrrole αCH2 bonds are in the intermediate exchange regime. As can be seen, most (ΔM0)NP2 samples showed the presence of low-spin impurities that could not be removed from these small samples. The A and B heme orientations appear to have rather different H57 orientations in some mutants (I120T, F42A, Y104A, Y85A), as is seen not only by the difference in chemical shift between the resonances of methyls 5 and 8 for A and B, but also by the difference in histidine β-CH chemical shift (18–22 ppm) for B versus A, respectively. These differences suggest that the heme seating is somewhat different for A and B. All samples were maintained at room temperature for at least 8 days before the spectra were recorded to ensure that the equilibrium A to B ratio had been reached, and in some cases for considerably longer periods of time; the parent of this series, NP2(D1A), required equilibration for over 3 months to achieve that goal
Although most of the mutants prepared in this work in the NP2(D1A) and (ΔM0)NP2 series involve residues whose side chains interact directly with the edges of the heme (“belt” mutants), the NP2(D1A,I120T), (ΔM0)NP2(I120T), and NP2(D1A,F66A) mutant proteins were also prepared. It can be seen in Fig. 4 that for the I120T mutants of each N-terminus, the 1D 1H NMR spectra are also almost identical [except for the small impurity peaks of the (ΔM0) mutants], and these two mutants each have approximate 1:1 A to B heme orientation ratios, as compared with the 2:3 A to B heme orientation ratio of the same mutant of the M0 series [38]. However, assignment of the heme resonances of NP2(D1A,I120T) by STD spectroscopy (Fig. S2) showed that the heme seatings of A and B are actually fairly similar to those seen in previous mutants of the M0 series [38]: 5M and 8M of A are fairly close together (0.8 ppm), suggesting a H57 ligand plane orientation of about 133°, whereas 8M and 5M of B are even closer together, only 0.3 ppm apart, 8M of B at 59.5 ppm and 5M of B at 59.2 ppm at 30 °C, suggesting a H57 ligand plane orientation of a little more than 135° for B. We expect that these H57 imidazole plane orientation changes for A and B represent changes in heme seating, rather than changes in the angles of the side chain rotamer of H57, and thus in this case a clockwise rotation of the heme of about 2° for A, and a counterclockwise rotation of the heme by less than 1° for B as we look down at the heme from the distal pocket. Such a rotation, with the large I120 side chain gone, would relieve the short distance of 2.68 Å between the 4-vinyl-β-cis proton and the L55 Cδ2 methyl protons of the B heme orientation, and the much more important 2-vinyl-β-trans interaction with the phenyl ring of F42 of the A heme orientation. Both the difference in the A to B ratio and the seating differences for the A and B isomers are presumably a result of the A–B and G–H loops being able to close in the NP2(D1A) and (ΔM0)NP2 series, thus decreasing the size of the distal pocket in comparison with that of (M0)NP2 [38]. The H57 β-CH resonances at near 18 and 20 ppm for B and A, respectively, clearly confirm that the A to B ratio is close to 1:1. Although the two resonances have different heights and linewidths, their integrated ratios are essentially identical.
The following NP2(D1A) mutants were prepared to probe the importance of the belt of aromatic residues around the heme: NP2(D1A,Y38A), NP2(D1A,F42A), NP2(D1A,Y85A), NP2(D1A,Y104A), and, although it is not part of the belt, NP2(D1A,F66A), where F66 is beneath the heme, with the phenyl ring in a plane nearly parallel to that of the imidazole ring of H57, the protein-provided heme ligand. For (ΔM0)NP2, the following belt mutants were prepared for comparison with the NP2(D1A) belt mutants, some to probe the importance of the tyrosyl as compared with the phenyl side chain: (ΔM0)NP2(Y38F), (ΔM0)NP2(Y85F) (NP1 and NP4 have phenylalanine at this position), and (ΔM0)NP2(Y104A). As mentioned in “Materials and methods,” although both the (ΔM0)NP2(Y104F) gene and the NP4(Y105F) gene were created and expressed, no correctly folded protein could be obtained in either case. This was also found to be the case for (M0)NP2(Y104F) and NP2(D1A,Y104F) (A.M. Goren, unpublished results). Thus, loss of the hydrogen bond between Y104(105) and the E53(55) carboxyl, shown in an earlier publication [37] and reprinted in Fig. S4, has a significant destabilizing effect on the protein. However, replacement of Y104(105) with the much smaller alanine did produce correctly folded proteins. The fact that these tyrosine to alanine mutant proteins folded and incorporated heme suggests that the large cavity created by removal of the tyrosine side chain allows space for multiple water molecules to enter the calyx and stabilize the charge on the carboxylate of E53 at pH values above its pK a (approximately 5.8 [25]).
For the NP2(D1A,F66A) mutant, the A to B ratio is similar to that of the parent, of the order of 1:14, and there is only a small angular change in the heme seating of about 2°. The H57 β-CH resonance is at slightly below 20 ppm, also consistent with the B heme orientation. In comparison, although the spectra have some similarities with regard to the pattern of heme methyl resonances, the ones in which the belt is Y104A and Y85A were quite difficult to assign to A or B. The larger chemical shift ranges of the NP2(D1A,Y104A), (ΔM0)NP2(Y104A), and NP2(D1A,Y85A) mutants suggested to us early in our study that the major heme orientation isomer was A, as did the H57 β-CH chemical shift of 21 ppm or greater, whereas the small peak at about 75 ppm of each spectrum suggested that it represented the 5-methyl and 8-methyl resonances of the minor A heme orientation, whereas the major isomer was B. In addition, the larger chemical shift range of 21 ppm or larger for the heme methyl resonances could be simply a result of the removal of the tyrosine –OH. We had seen decreases in chemical shift ranges among a number of mutants where a thronine –OH was substituted for a methyl group of leucine or valine, and the reverse where a serine –OH was replaced by a methyl of alanine [38]. However, in the tyrosine to alanine mutants of this study, the change is in the reverse direction. But it is likely that the removal of the large protein side chain leaves extra space which could be filled by water molecules, thus possibly reversing the trend seen previously [38]. Because of the ambiguity in heme orientation, 1D STD experiments were conducted to assign the heme methyls of NP2(D1A,Y104A). It was found, as shown in Fig. S6, that the high-spin heme methyl order is 5M > 8M > 1M > 3M, which has, throughout this study, been diagnostic of the A heme orientation. However, assignment of the natural abundance 1H{13C} HMQC and NOESY spectra of the low-spin NMeIm complex of NP2(D1A,Y104A), shown in Fig. S5, was consistent only with it being the B isomer, since there are clear nuclear Overhauser effect cross peaks from I120 CδH3 to 3M and 2Hα, and from L132 CδH3 to 2Hα, which are only possible for the B heme orientation. Thus, the STD data alone might have convinced us that the major orientation is A, but the nuclear Overhauser effect cross peaks for the low-spin NMeIm complex confirm that the major orientation is actually B, and thus the 5M > 8M > 1M > 3M order is a special case for this mutant and the Y85A mutant. Because of the similarity in the chemical shifts of (ΔM0)NP2(Y104A) and those of NP2(D1A,Y104A), we can be sure that B is the major heme orientation for that mutant as well. And because of the very similar spectral pattern for NP2(D1A,Y85A), it is reasonable to assume that this mutant also has B as the major heme orientation, although we have not investigated it in as much detail.
An interesting finding for the high-spin Y104A mutants is that the 4Hα chemical shift is 55.2 ppm, as shown in Fig. 5, which summarizes the information obtained from the STD data in Fig. S5. This value, 55.2 ppm, is the highest value of the chemical shift for 4Hα observed thus far for the nitrophorins. It is higher, by far, than the chemical shift of 2Hα, 38.2 ppm, and thus tells us that there is significantly more spin density at the 4-α-vinyl carbon. We see this effect for the Y104A and Y85A mutants, and for the triple mutant of NP2(D1A) discussed below, but in that case, for the 2-vinyl group. These mutants thus give us an idea as to the importance of vinyl in-plane or out-of-plane position with respect to the heme ring.

1D 1H NMR spectrum of NP2(D1A,Y104A) at 25 °C, showing the assignments of resonances from Fig. S6. By process of elimination, the 7Hα CH2 resonances must be those at 49.5 and 25.5 ppm. Note that at 25 °C the existence of two broad peaks at 50.5 and 49.5 ppm and two somewhat sharper peaks at 27 and 25.5 ppm is much more evident than in the saturation transfer/nuclear Overhauser effect difference spectra in Fig. S5, which were recorded at 30 °C. The closeness of each of these two 7Hα peaks to those of 6Hα probably means that both resonances were irradiated and showed difference spectra in Fig. S6. The small resonances at 74.8 and 62.8 ppm, and possibly at 41.7 and 22.1 ppm, are from the minor A heme orientation
For the major B heme orientations of NP2(D1A,Y104A) and NP2(D1A,Y85A), the heme seating is changed such that there is a 3–4-ppm difference in chemical shift for 5M and 8M, probably indicating a change in the heme seating of about 2° for the major B orientation of the Y104A mutants, and 3° for the Y85A mutant, in both cases by rotation of the heme in a clockwise direction, rather than the usually observed counterclockwise direction for the B heme orientation. Removal of the large phenol side chain of Y104 probably allows E53 to change its position inside the calyx (see Fig. S4), thus creating additional space for the 4-vinyl group that causes the small changes in heme seating observed for these mutants, yet leaves the A to B heme orientation ratio unchanged from that of the NP2(D1A) or (ΔM0)NP2 parent. Thus, replacing the large phenol side chain of tyrosine with the methyl group of alanine creates additional space within the calyx which retains the favored B heme orientation, albeit with a slightly modified heme seating. However, since Y104(105) and Y85(F86) are common to all four adult Rhodnius nitrophorins, the properties of these mutants tell us nothing about the relative heme orientation stabilities of the NP1/NP4 versus the NP2/NP3 pairs.
For Y38F as Belt, the heme seating is similar to that of the (ΔM0)NP2 parent, as shown in Fig. 4 for (ΔM0)NP2 and (ΔM0)NP2(Y38F). In an earlier study of the peroxidase activity of (M0)NP2, (M0)NP2(Y38F) was prepared, and when treated with hydrogen peroxide or peroxyacetic acid, it gave rise to a different tyrosyl radical decomposition product than did (M0)NP2 itself [66]. This radical was shown to be located on Y85, by the fact that essentially no radical product was formed when the (M0)NP2(Y38F,Y85F) double mutant was reacted with hydrogen peroxide or peroxyacetic acid [66].
For Y38A as Belt, the heme seating is changed significantly, with both A and B having seatings different from those expected for a 135° angle for the H57 imidazole plane orientation, angles that are smaller than 135° for A and larger than 135° for B. In addition, the A to B ratio is close to 1:1, and there is little or no dynamics observed. At well over 4 Å for the four carbons of the phenol ring of Y38 closest to the heme, it is too far below the heme to form a π complex in the region of pyrrole ring IV, with 8M and 7P being the nearest substituents. The major difference in the NMR spectra of NP2(D1A,Y38A) and (ΔM0)NP2(Y38F) suggests that for the Y38A mutant, the β-barrel is able to collapse toward the heme to take up much of the space lost by removal of the tyrosine side chain, thus changing the NMR spectrum markedly. In any case, this is a second case, after the I120T mutant, where the extra stability of the B heme orientation of NP2 is lost. However, since Y38(40) is common to all four nitrophorins of the adult insect, this mutant tells us nothing about the differences in heme orientation stability between the NP1/NP4 and NP2/NP3 pairs of proteins.
The NP2(D1A,Belt) mutant in which Belt is F42A shows a large predominance of the A heme orientation, A to B ratio of approximately 10:1, but with the seating of both A and B being very close to that expected for the axial ligand plane oriented at exactly 135°. In support of the assignment of A as the major isomer, we note that the two overlapped methyl resonances at 68 ppm are at a chemical shift very similar to that of the small methyl resonance of the parent NP2(D1A) complex (Fig. 4), whereas the small resonance at approximately 64.5 ppm is at a chemical shift very similar to that of the large resonance of the two overlapped methyl resonances at 65 ppm. At the same time, the small, broad resonance at 54 ppm for NP2(D1A) is at a chemical shift very similar to that of the large methyl resonance of NP2(D1A,F42A) assigned to the 3M group of that mutant. We also note that the large H57 β-CH resonance is at a higher chemical shift, 21.5 ppm, with the small resonance being at 20.0 ppm. This order is opposite that found in all cases of (M0)NP2 mutants where separate H57 β-CH resonances have been observed, in which the B heme orientation is more abundant than the A heme orientation [38]. Assignment of the heme resonances of high-spin NP2(D1A,F42A) is shown in Fig. 6. This material comes from the assignment of the low-spin NMeIm complex (Fig. S7) and the STD spectra which connect the low-spin assignments to the high-spin assignments (Fig. S8). Thus, we see that the mutation F42A makes the A heme orientation very highly favored, the only single mutant of (M0)NP2 or NP2(D1A) besides (M0)NP2(V24E) [37, 67, 68] in which the A heme orientation is favored, and, as was the case in that former study, mutation of only one residue is required to create this opposite preference for the heme orientation. Thus, position 42 of NP2 is extremely important in determining the A to B heme orientation ratio.

Mutants aimed at recreating, in a stepwise manner, the heme environment of NP4, and their 1H NMR spectra in the high-spin Fe(III) state. For NP2(D1A,I120T) the methyl resonances of A are identified in blue lettering, whereas those of B are given in red lettering
Positions 42 and 106 for NP2 and the corresponding positions 44 and 107 for NP1 and NP4 are quite interesting in that, as shown in Fig. 1, there are residues at positions 42/44 and at 106/107 which are the reciprocal of each other, with the 42/44 residue being mainly below and the 106/107 residue being mainly above the mean plane of the heme. NP2 has F42 and L106, whereas NP1 and NP4 have L44 and F107; in each case the larger phenylalanine is paired with the smaller leucine, as shown in Fig. 1, such that the amount of space used by the combination would seem to be fairly constant with respect to their effect on the orientation of the heme. We have already prepared and investigated in detail one of the two noncomplementary mutants, NP1(L44F), in which two phenyl groups are facing and probably interfering with each other [38]. The 1H NMR spectrum of the high-spin form of the noncomplementary mutant NP1(L44F) was found to exhibit chemical exchange among the heme methyl resonances and among the propionate α-CH resonances [36, 38]. Thus, it appeared that both of the heme orientations, while present, cannot achieve stable seatings with the heme in either A or B orientation in this mutant, and were each changing their heme seating on the NMR timescale. Therefore, we did not prepare the other noncomplementary mutant, NP2(L106F), either previously or in this work.
We did, however, prepare a triple mutant of NP2(D1A), which was designed to mimic the heme pocket of NP1, or better (because of the N-terminus), that of NP4, both of which have approximately 1:1 equilibrium ratios of A and B. The 1D 1H NMR spectrum of this high-spin triple mutant, the aqua complex of NP2(D1A,F42L,L106F,I120T), is included in Fig. 4, and is also shown, together with the spectra of NP2(D1A), NP2(D1A,F42A), and NP2(D1A,I120T) in Fig. 6, which shows the changes in the spectra with each mutation, albeit F42L in the triple mutant of NP2(D1A). By detailed investigation of this triple mutant of NP2(D1A), described below, as both the high-spin and the low-spin complex, we have confirmed that the heme orientation is very nearly all A, about 25:1 A to B, unlike the high-spin wild-type NP4 aqua complex, which has an A to B ratio of approximately 1:1. Thus, this NP2(D1A) triple mutant has far overcorrected for the preference of the B heme orientation of NP2(D1A), an approximate change in the equilibrium constant from 1:14 to 25:1, a factor of 350. This overcompensation is likely a result of the fact that the fit of the heme inside NP2 is much tighter than that inside NP4 or NP1, as can be seen by the much severer ruffling of the heme for NP2 as compared with NP4 or NP1, as shown in Table S1. Thus, when the residues native to NP2 are replaced by those of the smaller side chains present in NP4, the result is a strong overcompensation and stabilization of the A heme orientation over the B heme orientation.
The heme resonances of the high-spin aqua complex of NP2(D1A,F42L,L106F,I120T) were assigned via saturation transfer experiments (shown in Figs. S10, S11) performed on solutions which contained about 0.25 equiv NMeIm, using the assignments of the low-spin NMeIm complex shown in Fig. S9. In addition, a WEFT-NOESY spectrum confirmed the α-CH2 assignments of the 6- and 7-propionates of the pure high-spin complex (Fig. S12). Unique features of the 1D 1H NMR spectrum of the high-spin form of this mutant include the following: (a) 2-vinyl-α-H, at 55.2 ppm, has a larger chemical shift than 3M, and the same chemical shift as 4-vinyl in NP2(D1A,Y104A) discussed above; (b) 4-vinyl-α-H has a larger chemical shift (42.5 ppm) than in many other A heme orientation NP2 mutants; (c) the H57 β-CH resonance at 20.2 ppm is accompanied by a very small resonance at about 18.8 ppm that arises from the B heme orientation. The chemical shifts of the heme substituents of each of the high-spin proteins discussed in this section are presented in Table 1.
NMR characterization of the low-spin ligand complexes of NP2(D1A) belt mutants
The 1D and WEFT-NOESY spectra of NP2(D1A,I120T)-NMeIm, NP2(D1A,Y104A)-NMeIm, NP2(D1A,F42A)-NMeIm, and NP2(D1A,F42L,L106F,I120T)-NMeIm are shown in Figs. S2, S5, S7, and S9, respectively. As can be seen, two of these mutants show predominantly the A heme orientation, with ratios of A to B of about 10:1 and 25:1, respectively, for NP2(D1A,F42A)-NMeIm and NP2(D1A,F42L,L106F,I120T)-NMeIm. The most difficult spectrum to assign was that of NP2(D1A,I120T)-NMeIm, because of having both A and B heme orientations present in equal abundance. Although the 2Hα proton of B could be assigned, the 2Hα proton of A eluded assignment. The reverse was true for the 4Hα proton, where 4Hα and 4Hβ of A could be assigned but 4Hα and 4Hβ of B could not.
For NP2(D1A,Y104A)-NMeIm, for which the HMQC and WEFT-NOESY spectra are shown in Fig. S5, we see that, as for the high-spin form of this mutant, the 4Hα resonance is at an unusual chemical shift of 23.7 ppm, whereas it is usually found at about 6 ppm for the B heme orientation [6.4 ppm for NP2(D1A)-NMeIm, Table 2] [35]. The much larger chemical shift of 4Hα for the Y104A mutants of NP2(D1A) and (ΔM0)NP2 suggests a much larger spin density at that position, which, for the B heme orientation, is right at the site of the Y104A mutation. The very strong nuclear Overhauser effect cross peaks between 4Hα and 5M and between 4Hβ at 0.47 ppm and 3M, however, confirm that the vinyl group is still positioned more or less as seen in Fig. S4, with 4Hα pointing toward 5M, albeit with the vinyl group fully in the plane of the heme. The chemical shifts and assignments of all the complexes are presented in Tables 2 and 3, where the parent complex, NP2(D1A)-NMeIm [35], is also included in Table 2.
For the low-spin complex of the triple mutant of NP2(D1A) bound to NMeIm, the complex exhibits one major heme orientation, that being the A orientation, with A to B ratio of approximately 25:1, as shown in Fig. S9, as compared with 1:14 for NP2(D1A), a change in K eq from 0.071 to 25, a factor of 350. The heme of the A orientation rotates clockwise by 2–3° to remove 2-vinyl from being near L42 and F106, and this 2-vinyl has its Hα resonance at the highest chemical shift observed thus far for this heme substituent in any NP2 mutant (55 ppm), a larger chemical shift than for 3M (52.3 ppm). For NP2(D1A) the 2Hα chemical shift is 44.8 ppm, and for most other mutants in Fig. 4 it is at 42–45 ppm, at 45.3 ppm for NP2(D1A,F42A), and at 55.2 ppm for NP2(D1A,F42L,L106F,I120T), whereas for NP2(D1A,Y104A) and (ΔM0)NP2(Y104A) it is at 38.2 ppm and it is at 38.0 ppm for NP2(D1A,Y85A). In contrast, 4Hα, which is usually at 33–41 ppm, is found at 55.2 ppm for NP2(D1A,Y104A) and (ΔM0)NP2(Y104A), and fairly close to 51.7 ppm for NP2(D1A,Y85A), suggesting that removal of the large tyrosyl side chains of each of these mutants forces the 4-vinyl group to be in much greater conjugation with the heme π system of the high-spin complexes. The reverse is true for the triple mutant of NP2(D1A), which has A heme orientation and has 2Hα at 55.2 ppm and 4Hα at 42.5 ppm. Since the triple mutant of NP2(D1A) has major heme orientation A, whereas the Y104A and Y85A mutants have heme orientation B, it is unlikely that the chemical shifts of 2Hα or 4Hα are related to heme orientation. Rather, a larger chemical shift for either vinyl Hα is consistent with greater spin delocalization to that vinyl group, which is consistent with a more in-plane position of the vinyl side chain. Since being coplanar with the heme is a higher-energy state for the vinyls of protohemin, something must be forcing the 4-vinyl of Y104A mutants to be coplanar with the heme. Without a crystal structure of this mutant it is difficult to determine which protein side chain is involved, since it is clear that there could have been some major changes in packing of the side chains near 4-vinyl and 3-methyl in these mutants.
The NMeIm complex of NP2(D1A,F42L,L106F,I120T) has an extremely well resolved NMR spectrum that does not exhibit any chemical exchange derived from two different environments for a particular resonance. Three well-resolved protein methyl resonances are observed at negative chemical shifts in the natural abundance 1H{13C} HMQC spectrum, at 1H chemical shifts of −0.7, −1.3, and −2.8 ppm, all with 13C shifts of approximately 20 ppm, as seen in Fig. S9. The first two likely arise from the Cδ1 and Cδ2 methyls of L132, both of which give cross peaks to 3M, but the third has not been assigned.
Summary and conclusions
The NP2 proteins with N-termini ΔM0 and D1A continue to show extremely similar 1D and 2D NMR spectra, as reported previously [40], and can be investigated interchangeably. I120 δ-CH3 is very close to the heme, and together with the cavity structure defines the heme orientation (A to B) ratio inside the heme cavity, so the I120T mutants of (ΔM0)NP2 and NP2(D1A) provide essentially equal stability for the A and B heme orientations. The mutation F42A reverses the heme orientation to an A to B ratio of approximately 10:1. The triple mutant of NP2(D1A) vastly overcorrects for the approximately equal stability of the A and B orientations of the I120T mutants and creates an A to B ratio of approximately 25:1. Finally, removal of a phenol side chain of (ΔM0)NP2 or NP2(D1A) from Y104 or Y85, replacing it with a proton (methyl of alanine vs CH2 of tyrosine), or by creating the NP2(D1A) triple mutant, allows 4Hα for B of Y104A mutants or 2Hα for A of the triple mutant to have a larger chemical shift than 3M, suggesting that the vinyl group is in as full conjugation with the heme π system as possible. Thus, the effects of the vinyl groups, with just two more carbons than would be present in the “symmetrical hemin” (2,4-dimethyldeuterohemin),have a number of important effects on the NMR spectra of NP2, where the heme seems to fit more tightly into the calyx than is true of NP1 and NP4.
References
Walker FA (2005) J Inorg Biochem 99:216–236
Walker FA, Montfort WR (2001) Adv Inorg Chem 51:295–358
Ribeiro JMC, Hazzard JMH, Nussenzveig RH, Champagne DE, Walker FA (1993) Science 260:539–541
Yuda M, Hirai M, Miura K, Matsumura H, Ando K, Chinzei Y (1996) Eur J Biochem 242:807–812
Ribeiro JMC, Nussenzveig RH (1993) FEBS Lett 330:165–168
Nussenzveig RH, Bentley DL, Ribeiro JMC (1995) J Exp Biol 198:1093–1098
Ribeiro JMC, Walker FA (1994) J Exp Med 180:2251–2258
Ribeiro JMC, Schneider M, Guimaraes JA (1995) Biochem J 308:243–249
Zhang Y, Ribeiro JMC, Guimaraes JA, Walsh PN (1998) Biochemistry 37:10681–10690
Mizurini DM, Francischetti IM, Andersen JF, Monteiro RQ (2010) Thromb Haemost 104:1116–1123
Weichsel A, Andersen JF, Champagne DE, Walker FA, Montfort WR (1998) Nat Struct Biol 5:304–309
Ding XD, Weichsel A, Andersen JF, Shokhireva TK, Balfour C, Pierik AJ, Averill BA, Montfort WR, Walker FA (1999) J Am Chem Soc 121:128–138
Vetter SW, Terentis AC, Osborne RL, Dawson JH, Goodin DB (2009) J Biol Inorg Chem 14:179–191
Andersen JF, Montfort WR (2000) J Biol Chem 275:30496–30503
Weichsel A, Berry RE, Zhang H, Walker FA, Montfort WR (2005–2011) Crystal structures, ligand induced conformational change and heme deformation in complexes of nitrophorin 2, a nitric oxide transport protein from Rhodnius prolixus. http://www.rcsb.org/pdb/explore/explore.do?structureId=2EU7)
Shokhireva TK, Weichsel A, Smith KM, Berry RE, Shokhirev NV, Balfour CA, Zhang H, Montfort WR, Walker FA (2007) Inorg Chem 46:2041–2056
Andersen JF, Weichsel A, Balfour CA, Champagne DE, Montfort WR (1998) Structure 6:1315–1327
Weichsel A, Andersen JF, Roberts SA, Montfort WR (2000) Nat Struct Biol 7:551–554
Roberts SA, Weichsel A, Qiu Y, Shelnutt JA, Walker FA, Montfort WR (2001) Biochemistry 40:11327–11337
Maes EM, Weichsel A, Andersen JF, Shepley D, Montfort WR (2004) Biochemistry 43:6679–6690
Maes EM, Roberts SA, Weichsel A, Montfort WR (2005) Biochemistry 44:12690–12699
Kondrashov DA, Roberts SA, Weichsel A, Montfort WR (2004) Biochemistry 43:13637–13647
He C, Ogata H, Knipp M (2010) Biochemistry 49:5841–5851
Berry RE, Ding XD, Shokhireva TK, Weichsel A, Montfort WR, Walker FA (2004) J Biol Inorg Chem 9:135–144
Berry RE, Shokhirev MN, Ho AYW, Yang F, Shokhireva TK, Zhang H, Weichsel A, Montfort WR, Walker FA (2009) J Am Chem Soc 131:2313–2327
Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM (1997) Structure 5:1093–1108
Smith LJ, Kahraman A, Thornton JM (2010) Proteins 78:2349–2368
Nienhaus K, Maes EM, Weichsel A, Montfort WR, Nienhaus GU (2004) J Biol Chem 279:39401–39407
Maes EM, Walker FA, Montfort WR, Czernuszewicz RS (2001) J Am Chem Soc 123:11664–11672
Astashkin AV, Raitsimring AM, Walker FA (1999) Chem Phys Lett 306:9–17
Shokhireva TK, Shokhirev NV, Walker FA (2003) Biochemistry 42:679–693
Shokhireva TK, Berry RE, Uno E, Balfour CA, Zhang H, Walker FA (2003) Proc Natl Acad Sci USA 100:3778–3783
Shokhireva TK, Smith KM, Berry RE, Shokhirev NV, Balfour CA, Zhang H, Walker FA (2007) Inorg Chem 46:170–178
Shokhireva TK, Berry RE, Zhang H, Shokhirev NV, Walker FA (2008) Inorg Chim Acta 361:925–940
Berry RE, Shokhireva TK, Filippov I, Shokhirev MN, Zhang H, Walker FA (2007) Biochemistry 46:6830–6843
Shokhireva TK, Shokhirev NV, Berry RE, Zhang H, Walker FA (2008) J Biol Inorg Chem 13:941–959
Yang F, Knipp M, Berry RE, Shokhireva TK, Zhang H, Walker FA (2009) J Biol Inorg Chem 14:1077–1095
Shokhireva TK, Berry RE, Zhang H, Walker FA (2011) J Inorg Biochem 105:1238–1257
Shokhireva TK, Walker FA (2012) J Biol Inorg Chem 17:911–926; erratum 1303
Berry RE, Muthu D, Shokhireva TK, Garrett SA, Zhang H, Walker FA (2012) Chem Biodiv 9:1739–1755
Abriata LA, Zaballa M-E, Berry RE, Yang F, Zhang H, Walker FA, Vila AJ (2013) Inorg Chem 52:1285–1295
Schünemann V, Benda R, Trautwein AX, Walker FA (2000) Isr J Chem 40:9–14
Moeser B, Janoschka A, Wolny JA, Paulsen H, Filippov I, Berry RE, Zhang H, Chumakov AI, Walker FA, Schünemann V (2012) J Am Chem Soc 134:4216–4228
Andersen JF, Ding XD, Balfour C, Shokhireva TK, Champagne DE, Walker FA, Montfort WR (2000) Biochemistry 39:10118–10131
Benabbas A, Ye X, Kubo M, Zhang Z, Maes EM, Montfort WR, Champion PM (2010) J Am Chem Soc 132:2811–2820
Abruzzetti S, He C, Ogata H, Bruno S, Viappiani C, Knipp M (2012) J Am Chem Soc 134:9986–9998
Hoshino M, Maeda M, Konishi R, Seki H, Ford PC (1996) J Am Chem Soc 118:5702–5707
Muthu D, Berry RE, Zhang H, Walker FA (2013) Biochemistry 52:7910–7925
Markley JL, Bax A, Arata Y, Hilbers CW, Kaptein R, Sykes BD, Wright PE, Wüthrich K (1998) J Biomol NMR 12:1–23
Shokhirev NV, Walker FA (1998) J Biol Inorg Chem 3:581–594
Zhong L, Wen X, Rabinowitz TM, Russell BS, Karan EF, Bren KL (2004) Proc Natl Acad Sci USA 101:8637–8642
Wen X, Bren KL (2005) Inorg Chem 44:8587–8593
Walker FA (2000) In: Kadish KM, Smith KM, Guilard R (eds) The porphyrin handbook, vol 5. Academic, Boston, pp 81–183
Walker FA (2010) In: Kadish KM, Smith KM, Guilard R (eds) The handbook of porphyrin science, vol VI. World Scientific, Hackensack, pp 1–337
Shokhirev NV, Walker FA (2011) ShiftPatterns. Available via http://www.shokhirev.com/nikolai/prgsciedu.html
Caignan GA, Deshmukh R, Wilks A, Zeng Y, Huang H, Moënne-Loccoz P, Bunce RA, Eastman MA, Rivera M (2002) J Am Chem Soc 124:14879–14892
Bertini I, Luchinat C, Parigi G, Walker FA (1999) J Biol Inorg Chem 4:515–519; erratum 846
Minniear AB, Shokhireva TK, Walker FA (1999) Inorg Chem 38:5856–5859
Caillet-Saguy C, Piccioli M, Turano P, Lukat-Rodgers G, Wolff N, Rodgers KR, Izadi-Pruneyre N, Delepierre M, Lecroisey A (2012) J Biol Chem 287:26932–26943
Caillet-Saguy C, Piccioli M, Turano P, Lukat-Rodgers G, Wolff N, Rodgers KR, Izadi-Pruneyre N, Delepierre M, Lecroisey A (2013) J Biol Chem 288:26942 (erratum)
Case DA (1995) J Biomol NMR 6:341–346
Haigh CW, Mallion RB (1980) Prog NMR Spectrosc 13:303–344
Johnson CE, Bovey FA (1958) J Chem Phys 29:1012–1014
Yang F, Shokhireva TK, Walker FA (2011) Inorg Chem 50:1176–1183
Barbush M, Dixon DW (1985) Biochem Biophys Res Commun 129:70–75
Singh R, Berry RE, Yang F, Zhang H, Walker FA, Ivancich A (2010) Biochemistry 49:8857–8872
Knipp M, Yang F, Berry RE, Zhang H, Shokhirev MN, Walker FA (2007) Biochemistry 46:13254–13268
Yang F, Zhang H, Knipp M (2009) Biochemistry 48:235–241
Acknowledgments
This article is dedicated to the memory of Professor Ivano Bertini in recognition of his many contributions to inorganic chemistry, bioinorganic chemistry, multidimensional NMR spectroscopy, and to the Society for Biological Inorganic Chemistry and this journal, the Journal of Biological Inorganic Chemistry. Professor Bertini’s role in the development of all of these areas and many others and his role as a mentor and friend are priceless; he is very much missed. This research was supported by the National Institutes of Health (grant HL054826), and the National Science Foundation (grant CHE-0809591). A.M.G. thanks the Undergraduate Biology Research Program and Biomedical Research Abroad: Vistas Open! for financial support.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Responsible editors: Lucia Banci, Claudio Luchinat.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Berry, R.E., Muthu, D., Shokhireva, T.K. et al. NMR investigations of nitrophorin 2 belt side chain effects on heme orientation and seating of native N-terminus NP2 and NP2(D1A). J Biol Inorg Chem 19, 577–593 (2014). https://doi.org/10.1007/s00775-013-1063-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-013-1063-8