
Abstract
The synthesis of new copper(II) bis(thiosemicarbazonato) complexes with an appended pyrene chromophore and their zinc(II) analogues is reported. The new proligands and their copper(II) and zinc(II) complexes were characterised by a combination of NMR, EPR, high performance liquid chromatography, mass spectrometry, electronic spectroscopy and electrochemical measurements. The new copper(II) complexes are fluorescent as a consequence of an appended pyrene substituent that is separated from the sulphur coordinating to the metal ion by five bonds. The emission from the pyrene substituent is concentration- and solvent-dependent with characteristic formation of excimer aggregates. A radioactive 64Cu complex has been prepared. Cell permeability, intracellular distribution and importantly the ability to cross the nuclear membrane to target DNA were investigated using confocal fluorescence microscopy in a human cancer cell line under normal oxygen conditions and hypoxic conditions. In both cases, there was no evidence of uptake of the copper(II) bis(thiosemicarbazonato) complexes in the area of the cell nucleus.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
There are several isotopes of copper that are of interest in the development of radiopharmaceuticals. Positron- and beta-emitting isotopes of copper with favourable decay characteristics are available, providing possibilities in both imaging and therapy [1, 2]. For example copper-64 decays via electron capture, positron, beta and Auger emissions; therefore, it can be used for both positron emission tomography and radiotherapeutic applications. Low dioxygen concentration (hypoxia) has emerged as an important factor in tumour biology. Hypoxic tissue is associated with advanced solid tumours as the dioxygen consumption rate of the cancerous cells exceeds supply [3]. Correlations exist between hypoxia, tumour aggressiveness, angiogenesis and response to treatment. Consequently, the development of hypoxia-selective imaging agents would help to guide therapeutic intervention and also the use of therapeutic copper isotopes offers the potential for hypoxia-targeted radiotherapy.
Development of targeted copper radiopharmaceuticals requires suitable stable chelators. Radioactive copper complexes with bis(thiosemicarbazone) ligands have been investigated as perfusion tracers and as hypoxia-imaging agents. The complexes are stable, neutral, lipophilic and cell-permeable. The biodistribution of copper bis(thiosemicarbazonato) complexes is remarkably sensitive to the nature of the backbone substituents on the diimine backbone (Fig. 1) [4–6]. The copper complex formed from the ligand with two methyl substituents on the backbone—diacetyl bis(N 4-methylthiosemicarbazonato)copper(II), CuII(atsm) (Fig. 1)—is selectively retained in hypoxic cells. The hypoxia selectivity of CuII(atsm) has been attributed to a reductive trapping mechanism involving the reduction of the metal from CuII to CuI [4, 7–13]. On the other hand, glyoxal bis(N 4-methylthiosemicarbazonato)copper(II), CuII(gtsm) (Fig. 1) releases copper intracellularly inside all cells and is not selective for hypoxic cells [6, 14–16]. CuII(atsm) hypoxia selectivity has been correlated with the CuII/CuI reduction potential as CuII(atsm) is more difficult to reduce than CuII(gtsm) by some 160 mV [12, 15, 17].

Diacetyl bis(N 4-methylthiosemicarbazonato)copper(II), CuII(atsm), R1 and R2 are CH3; glyoxal bis(N 4-methylthiosemicarbazonato)copper(II), CuII(gtsm), R1 is H, R2 is CH3
Little is known about the mechanisms of uptake of bis(thiosemicarbazonato)copper(II) complexes or their intracellular distribution, but recent studies have highlighted cell-line-dependent differences in uptake and retention [18]. Subcellular fractionation experiments were used to demonstrate the relative accumulation of radioactivity in cell nuclei, mitochondria and S2 fractions following treatment with radioactive bis(thiosemicarbazonato)copper(II) complexes [8, 19]. Zinc(II) bis(thiosemicarbazonato) complexes are weakly fluorescent owing to intraligand excitation and this fluorescence has been used to track the uptake of a range of complexes in a variety of cell lines using fluorescence microscopy [20]. The high resolution of fluorescence microscopy techniques offers potential advantages over detection methods based on radioactivity such as autoradiography. The analogous CuII complexes are not fluorescent. We have prepared fluorescent-tagged ligands, H2 L 1 and H2 L 2, by conjugating a pyrene fluorophore to the bifunctional ligands, H2atsm/a and H2atse/a. For fluorescence imaging inside cells it is desirable to use fluorophores that have relatively low energy excitation profiles to minimise ‘autofluorescence’ and cell damage. Pyrene has an excitation profile that is amenable to cellular imaging. Fluorescent labels can significantly alter the cell uptake and intracellular distribution when compared with ‘parent’ compounds, in this case CuII(atsm), but it was anticipated that the well-known DNA-intercalating properties of pyrene could be used to target the complex to cellular DNA to give a hypoxia-selective compound capable of binding to DNA [21, 22]. The prime target for therapeutic radiopharmaceuticals is the cell nucleus. The cytotoxicity of therapeutic radiopharmaceuticals results from modification of cellular DNA and subsequent apoptosis as a consequence of cellular changes initiated by ionising radiation [23]. The new copper(II) complexes are fluorescent as a consequence of an appended pyrene substituent that is separated from the sulphur coordinating to the metal ion by five bonds. Cell permeability, intracellular distribution and importantly the ability to cross the nuclear membrane to target DNA were investigated using confocal fluorescence microscopy in a human cancer cell line.
Results and discussion
Synthesis and characterisation
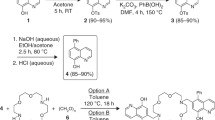
We recently reported the preparation of a new family of versatile bifunctional bis(thiosemicarbazone) proligands with a pendant amine (H2atsm/a and H2atse/a) suitable for conjugation to biomolecules [24–26]. New bis(thiosemicarbazone) proligands with an appended pyrene fluorophore, H2 L 1 and H2 L 2, were readily prepared by the reaction of H2atsm/a or H2atse/a with 1-pyrenecarboxaldehyde in ethanol (Scheme 1). Proligands of this type can exist in both E and Z isomeric forms via rotation about the C–C backbone and thiocarbohydrazone pyrene substituent. This can result in complicated temperature-dependent NMR. For example, the 1H NMR of H2 L 1 in deuterated dimethyl-d 6 sulphoxide (d 6-DMSO) reveals two broadened singlets at δ 2.26 and 2.31 ppm due to the methyl groups on the backbone of L 1H2 (N=C–CH3) with some evidence of fine structure. At 40 and 80 °C the signals are better resolved to give sharp singlets. At 40 °C the terminal methyl group of H2 L 1 (NH–CH3) gives rise to a doublet at δ 3.03 ppm (3 J HH = 5 Hz) as a consequence of coupling to the adjacent NH, which shifts downfield to δ ≈ 3.2 ppm at higher temperatures (80 °C) and is obscured by the peak due to H2O in the NMR solvent. On cooling, the spectrum is identical to that initially obtained at room temperature. The aromatic protons of the pyrene substituent and 13C resonances were assigned with the aid of correlation spectroscopy, heteronuclear multiple quantum correlation, heteronuclear multiple bond correlation and distortionless enhanced polarization transfer techniques. The signals for the two carbon atoms of the methyl substituents are at δ 11.2 and 11.5 ppm in the 13C NMR spectrum, the resonance attributed to the terminal methyl substituent (NH–CH3) is at δ 31.1 ppm, the two imine carbon atoms of the backbone of the ligand (C=N) give signals at δ 147.6 and 147.7 ppm and the two C=S carbon atoms result in a single slightly broad resonance at 178.6 ppm. The electrospray mass spectrum of each new ligand gave peaks that correspond to the protonated ligands, at m/z = 474 for [H2 L 1 + H+]+ and m/z = 486 for [H2 L 2 + H+]+. The purity of each ligand was confirmed by the presence of a single peak in the reverse-phase high performance liquid chromatograpy (HPLC) chromatogram (R T = 18.9 min, HPLC method A).

The preparation of H2 L 1, H2 L 2 and their CuII and ZnII complexes
Yellow-orange zinc complexes and red-brown copper complexes were readily prepared by the addition of either copper or zinc acetate to ethanolic mixtures of the proligand and heating at reflux. The complexes can also be prepared at room temperature in either dimethylformamide (DMF) or dimethyl sulphoxide (DMSO.) Alternatively the copper complex, CuII L 1, can be prepared by transmetallation of the zinc complex, ZnII L 1, using copper acetate in DMF [12, 27]. All metal complexes were characterised by electrospray mass spectrometry, microanalysis and reverse-phase HPLC, and in each case it was revealed that a single species was present. The copper complexes, CuII L 1 and CuII L 2, give signals in the electrospray mass spectrum at m/z = 535.068 (simulated, m/z = 535.067) and m/z = 549.082 (simulated, m/z = 549.082), respectively, corresponding to the protonated cations with the expected isotope pattern. A wide range of bis(thiosemicarbazonato)copper(II) complexes have been characterised by X-ray crystallography. The copper is often in a distorted square planar environment with a N2S2 donor set with a 5-5-5 chelate ring system (Fig. 1) and it is expected that the metal ion in CuII L 1 and CuII L 2 is similarly coordinated [11, 24, 28–32]. The X-band electron paramagnetic resonance (EPR) spectrum of CuII L 1 in CH2Cl2 at room temperature is essentially identical to that of CuII(astm) in the same solvent (both compounds are only sparingly soluble in CH2Cl2, but their solubility is sufficient to obtain EPR spectra). The spectrum exhibits characteristic copper hyperfine structure and superhyperfine coupling from two equivalent nitrogen donor atoms (89 G, A N = 16 G) and g iso = 2.03 [6, 25, 33]. NMR spectra of the diamagnetic complexes ZnII L 1 and ZnII L 2 were consistent with the proposed structures and the shifts in signals between the ligand and the zinc complexes are small. It is difficult to detect the resonance due to the C–S carbon atoms in the 13C NMR spectra. For example, in ZnL 1 a single weak and broad resonance is detectable at δ 176.9 ppm [even with a high number of acquisitions on a concentrated sample with a relatively long delay (d 1)]. Previous structural studies of bis(thiosemicarbazonato)zinc(II) complexes revealed that the metal ion is typically five coordinate and approximately square pyramidal, with the base of the pyramid composed of an N2S2 donor set provided by the bis(thiosemicarbazonato) ligand and the apical position occupied by a solvent molecule, an anion or the sulphur from an adjacent molecule [11, 20, 24, 34, 35]. Given the precedents, it is likely that ZnL 1 and ZnL 2 are also five coordinate about the zinc with the apical position occupied by a solvent molecule.
Electronic spectroscopy
The UV–vis spectrum of CuL 1 in DMSO revealed an absorption associated with the appended pyrene chromophore at λmax = 413 nm (ε = 30,933 M−1cm−1) which tailed into an absorption attributed to the CuN2S2 chromophore at λmax = 496 nm (ε = 17,700 M−1cm−1). The fluorescence spectrum for CuL 1 showed that the appended pyrene retains its fluorescence properties even when CuII is coordinated to the ligand. Excitation of dilute solutions of CuL 1 (3 μM) at λmax = 350 nm resulted in emission at λmax = 386 nm and λmax = 406 nm characteristic of pyrene, with the latter broad peak tailing to past 500 nm (Fig. 2). This was confirmed by titration of a solution of CuII(CH3CO2)2·H2O into a solution of H2 L 1. No changes to the fluorescence spectrum were observed, but complex formation was confirmed by monitoring changes in the visible spectrum and the growth of the absorbance attributed to the CuN2S2 chromophore at λmax = 496 nm. The conjugated pyrene substituent is sufficiently remote from the paramagnetic metal ion such that no quenching occurs [36].

Fluorescence spectrum of 3 × 10−6 M CuII L 1 in neat DMSO, λex = 350 nm
The emission profile of CuII L 1 is solvent-dependent. A 3 μM solution in aqueous buffer [phosphate buffer (PB), pH 7.4 with 20% DMSO (v/v) to assist with solubility] has a distinctly different appearance from that of the spectrum recorded at the same concentration in neat DMSO. In aqueous systems, excimer emission dominates, with a broad emission centred at λmax = 464 nm (λex = 364 nm) as a consequence of the hydrophobic chromophores aggregating to minimise their exposure to water (Fig. 3) [37]. Excimers are a result of intermolecular aggregates forming as a result of π-stacking interactions of aromatic rings.

Fluorescence spectrum of 3 × 10−6 M CuII L 1 in aqueous buffer (20% v/v DMSO/PB)
The emission spectrum of ZnII L 1 (10 μM, λex = 350 nm) exhibited the expected peaks associated with the pyrene substituent at 487 and 580 nm, but also a weaker-intensity emission centred at 580 nm. This weaker lower-energy peak is attributed to emission from the ZnIIN2S2 chromophore of the bis(thiosemicarbazonato) ligand as a result of Förster resonance energy transfer between the pyrene substituent and the ZnIIN2S2 chromophore. The fluorescence excitation spectrum for emission at 580 nm in a 12 μM solution showed excitation maxima at both 394 and 490 nm.
Electrochemistry of CuII L 1
Cyclic voltammetry measurements of CuII L 1 (Fig. 4) in DMSO with a glassy carbon working electrode revealed that the complex underwent a reversible one-electron reduction attributed to a CuII to CuI process at E°′ = −0.50 V versus the saturated calomel electrode (SCE). The anodic/cathodic peak separation for the CuII/I couple is 105 mV (under the same conditions ∆E = 159 mV for ferrocene/ferrocenium). In comparison hypoxia-selective CuII(atsm) undergoes a reversible one-electron reduction at E°′ = −0.59 V under the same conditions, whereas non-selective CuII(gtsm) undergoes a chemically reversible reduction at E°′ = 0.43 V (vs. SCE).

Cyclic voltammogram of CuII L 1 in DMSO, 0.1 M (Et4N)(PF6). Potentials are quoted versus an internal ferrocene/ferrocenium couple taken as having E°′ = 0.54 V (vs.the saturated calomel electrode)
Stability studies and radiolabelling with 64Cu
For radiopharmaceutical applications it is essential that 64Cu remains bound to the targeting ligand in vivo for sufficient time to allow appropriate localisation in target tissue. CuII(atsm) is of sufficient stability for application in both diagnosis and therapy. It was of interest to ascertain the stability of the new derivatives with respect to loss of the metal ion from the chelate and the stability of the hydrazinoimine linker between the DNA-targeting pyrene fluorophore and the bis(thiosemicarbazonato) ligand. Glutathione is a low molecular weight cysteinyl tripeptide present in relatively high concentrations (0.5–10 mM) in the cytosol of cells that is capable of acting as a reductant and of binding to metal ions. CuII L 1 (10 μM) was incubated in an aqueous buffer [PB, pH 7.4, 20% (v/v) DMSO] in the presence of glutathione (100 μM). The stability of CuII L 1 was monitored by HPLC analysis. Over a period of 4 h there was little change in the appearance of the chromatogram, indicating that under these conditions CuII L 1 is stable to hydrolysis of the pyrene substituent and loss of the metal ion (more than 90% remained unchanged). The complex was also stable in a ‘cysteine and histidine challenge’ experiment in which CuII L 1 (10 μM) was incubated at 37 °C in the presence of histidine and cysteine (100 μM).
The 64Cu complex can be prepared via transmetallation of ZnL 1 to give 64CuL 1 in high radiochemical purity. 64CuCl2 was reacted with an excess of ZnL 1 in DMSO (1 mg/mL) and sodium acetate buffer solution. Analysis by radio-HPLC indicates 64CuL 1 (R T = 19.10 min) with a radiochemical purity of more than 95% (1.8 MBq/μg of ZnII L 1) (Fig. 5). Radiolabelling via transmetallation gave products of higher radiochemical purity than synthesis direct from the ‘free’ ligand, which is consistent with previous observations on related systems [25, 38].

Radio high performance liquid chromatography of 64CuII L 1 prepared by transmetallation from ZnII L 1; C18 reverse-phase column
Cell uptake and washout of CuIIL1
Cell uptake and washout was investigated by treating a cervical cancer cell line (HeLa) with CuII L 1 (total complex concentration 50 μM) and measuring the intracellular copper concentration by inductively coupled plasma mass spectrometry (ICP-MS) [16, 39, 40]. Intracellular copper concentrations for the treated cells were compared with those of an untreated control. Replacing the cell medium for treated cultures reverses the copper concentration gradient and effectively ‘washes out’ the complex from the cell. For this washout to occur, it is assumed that the complex retains its structural integrity inside the cell, that is, the CuII is not reduced to more labile CuI followed by dissociation from the bis(thisoemicarbazonato) ligand and sequestration by intracellular CuI binding proteins. Hypoxia-selective CuII(atsm) has ‘washout’ behaviour dramatically different from that of non-selective CuII(gtsm). Both compounds are cell-permeable, increasing the intracellular copper concentration. In the case of CuII(atsm) there is a 91 ± 19-fold increase in intracellular copper concentrations when compared with an untreated control, whereas treatment with CuII(gtsm) results in a 202 ± 18-fold increase. In the case of CuII(atsm), replacement of the medium after the initial treatment of the cells reduced the intracellular copper concentration to 40% of the value after treatment (37 ± 12-fold increase compared with an untreated control). In comparison with CuII(gtsm), incubating the cells in fresh medium had little effect; the copper is trapped inside the cell, presumably owing to reductive assisted transchelation (Fig. 6). In the case of CuII L 1, efficient cell permeability was indicated by the 85 ± 11-fold increase in the intracellular copper concentration when compared with an untreated control. Encouragingly, replacing the cell medium with fresh medium resulted in a significant reduction in the intracellular copper levels (reduced to a 8 ± 2-fold increase), and the effective ‘washout’ of the complex from the cells was analogous to that for hypoxia-selective CuII(atsm). These preliminary cell uptake and washout studies suggest that the new complex, CuII L 1, with a conjugated fluorophore, has cellular uptake and washout behaviour similar to that of hypoxia-selective CuII(atsm).

Intracellular copper concentrations in HeLa cell pellets following treatment with copper complexes (50 μM) measured by inductively coupled plasma mass spectrometry. Copper concentrations are plotted as the fold increase compared with cells not treated with copper complexes (untreated control, 0.064 μM copper)
Fluorescence studies of the intracellular distribution of CuL 1 in cancer cells
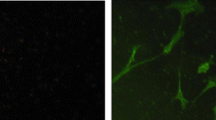
Fluorescence microscopy has been used to track the cell uptake and intracellular distribution of zinc(II) bis(thiosemicarbazonato) complexes that are weakly fluorescent owing to intraligand excitation [20]. The analogous copper(II) compounds are not fluorescent. The new systems with a conjugated pyrene fluorophore are fluorescent even in the presence of paramagnetic d 9 CuII. Cell uptake and the intracellular distribution of CuII L 1 in HeLa cells were determined by confocal fluorescence microscopy, first for a copper(II) bis(thiosemicarbazonato) complex. Differential interference contrast and confocal fluorescence microscopy images of HeLa cells treated with CuII L 1 (50 μM) and fluorescence microscopy images (λex = 405 nm, λex = 410–470 nm) are shown in Fig. 7. These measurements confirmed the cell-uptake data obtained by ICP-MS and showed that CuII L 1 is cell-permeable. There was no apparent cell toxicity at these concentrations as determined by a lactate dehydrogenase assay. The compound localises predominantly in the cytosol, with punctate inclusions, possibly vesicles, also observed within the cytosol. Importantly, considering the potential of pyrene derivatives to bind DNA, we saw no staining of the nucleus with CuII L 1. The lack of fluorescence in the nucleus does not completely preclude the possibility of uptake of the complex in the nucleus. In some cases the fluorophore interacts with DNA in such a way that the emission is quenched or significantly shifted. DNA has been shown to dampen the fluorescence of platinum complexes with an anthraquinone functional group and platinum complexes with a coumarin 120 fluorophore [41, 42]. In the present case, titration of calf thymus DNA into a buffered solution of CuII L 1 resulted in an initial small reduction in fluorescence intensity and a slight shift to a longer wavelength (λem = 464–468 nm), but further additions did not result in a linear reduction in fluorescence intensity and did not result in complete quenching (Fig. S1). The punctate vesicle-like structures where the compound localises are often relatively large and distinct, appearing to be morphologically consistent with autophagic vacuoles [43]. Autophagy is a cellular degradation system that is induced to remove and recycle cytoplasmic constituents, including organelles and foreign material. A double membrane is formed around the cytoplasmic constituents, forming autophagosomes, which subsequently fuse with lysosomes for degradation [44].

HeLa cells exposed to 50 μM CuII L 1 reveal the presence of cytoplasmic fluorescence, including both diffuse fluorescence and fluorescence from large vesicle-like structures. A proportion of the structures colocalise with LysoTracker Red, indicative of lysosomal and/or autophagic vacuole origin. DIC differential interference contrast image, LR LysoTracker Red, ToPro3 To-Pro-3 iodide, bar 10 μm
To investigate whether the punctate vesicle-like structures were undergoing lysosomal-associated degradation, we used LysoTracker Red (LR) and AO (data not shown), which label acidic compartments, including lysosomes and autophagic vacuoles. The vesicle-like structures revealed only partial colocalisation with LR (Fig. 7). The incomplete colocalisation may be explained by the possibility that some of the structures are early autophagosomes, and have yet to fuse with lysosomes and therefore cannot be detected by LR. Alternatively, the LR-negative structures may represent an alternative trafficking pathway for CuII L 1 and further studies will be needed to delineate these complex subcellular processes.
Hypoxia is known to alter intracellular retention of CuII(atsm), which has been described as a hypoxia-selective compound. We examined the intracellular distribution of CuII L 1 in hypoxic cells by the BD GasPak™ chamber system to induce hypoxia [45, 46]. HeLa cells were exposed to hypoxia for 6 h, treated with CuII L 1, then incubated for a further 30 min under hypoxia. The low-oxygen condition did not result in nuclear localisation of CuII L 1 (Fig. 8). Formation of vesicles was still observed under hypoxia and there was little colocalisation with the LR marker. Interestingly, the overlay also revealed that some of the larger structures may be on the cell surface, indicating a potential involvement in a secretory pathway for processing of the vesicles. Whether this is enhanced by the hypoxic conditions is not known. This may also contribute to altered morphology of the cells. These findings indicate that although hypoxia does not induce nuclear uptake of CuII L 1, it may still have substantial effects on the metabolism of the compound and this will need to be investigated further.

HeLa cells exposed to 50 μM CuII L 1 under hypoxia reveal the presence of cytoplasmic fluorescence, including both diffuse fluorescence and fluorescence from large vesicle-like structures as seen under normoxia in Fig. 7. Little colocalisation with LysoTracker Red was observed. No specific nuclear localisation was observed. LysoRed LysoTracker Red, bar 10 μm
Conclusion
The synthesis of new bis(thiosemicarbazonato)copper(II) complexes with an appended pyrene chromophore and their zinc analogues was reported. The new proligands and their copper(II) and zinc(II) complexes were characterised by a combination of NMR, EPR, HPLC, mass spectrometry, electronic spectroscopy and electrochemical measurements. The CuII/CuI couple is similar to that of the hypoxia-selective radiopharmaceuticals CuII(atsm). The new copper(II) complexes are fluorescent as a consequence of the appended pyrene substituent that is separated from the sulphur coordinating to the metal ion by five bonds. The emission from the pyrene substituent is concentration- and solvent-dependent, with characteristic formation of excimer aggregates. It is possible to prepare a radiolabelled 64Cu complex in high radiochemical purity via transmetallation from the zinc analogue. Cell permeability, intracellular distribution and importantly the ability to cross the nuclear membrane to target DNA were investigated using confocal fluorescence microscopy in a human cancer cell line coupled with analysis of copper levels with ICP-MS. Confocal microscopy confirmed the compound was taken up within HeLa cells and was distributed amongst the cytoplasm. Punctate vesicle-like structures were often relatively large and distinct, appearing to be morphologically consistent with autophagic vacuoles, although this is yet to be confirmed and alternative pathways of compound processing may be involved, such as a specific secretory pathway. A particular goal was to design a hypoxia-selective radiopharmaceutical that was targeted to nuclear DNA by a pyrene functional group, but there was no evidence of uptake in the area of the nucleus in the cell line investigated under normoxic or hypoxic conditions.
Materials and methods
General procedures
1H and 13C NMR spectra were recorded with an INOVA-500 spectromter (1H at 500 MHz and 13C at 125 MHz) and a Varian Unity 400 spectrometer (1H at 400 MHz and 13C at 100 MHz). Unless otherwise stated, all NMR spectra were recorded at room temperature. The reported chemical shifts (in parts per million) are referenced relative to residual solvent protons. Mass spectra were recorded using the electrospray technique (positive and negative ion) with a Micromass QUATTRO II triple-quadrapole electrospray mass spectrometer. EPR spectra were recorded with a Bruker FT ECS-106 spectrometer using 1,1-diphenyl-1,2-picrylhydrazyl as a reference.
Cyclic voltammograms were recorded with an AUTOLAB PGSTAT100 using the GPES V4.9 software program and employing a glassy carbon working electrode, a platinum counter electrode and a Ag/Ag+ reference electrode. The measurements were carried out in DMSO. The solutions contained approximately 4 mM analyte in 0.1 M tetraethylammonium hexafluorophosphate. The DMSO was dried over 3-Å molecular sieves under an atmosphere of N2 before use. Each solution was purged with N2 prior to analysis and was measured at ambient temperatures under a N2 atmosphere. Each sample was referenced to an internal reference of ferrocene, where the ferrocene/ferrocenium couple was taken as having E°′ = 0.54 V in DMSO versus the SCE.
Fluorescence excitation and emission spectra were measured in DMSO (0.01 mM) with a Varian Cary Eclipse fluorescence spectrophotometer with the Scan software program. UV–vis spectra were recorded in DMSO (0.01 mM) with a Shimadzu UV-1650PC UV–vis spectrophotometer using the UVPC c3.9 software program. HPLC was performed with an HP 1100 ChemStation system using a Supelco Discovery C18 column (150 mm × 4.6 mm, 5 μm). The HPLC solvents were 0.1% trifluoroacetic acid in water (solvent A) and 0.1% trifluoroacetic acid in acetonitrile (solvent B). The gradient was 0–100% solvent B in 0–25 min, the flow rate was 1.00 mL/min, and detection was at 280 nm. Thin-layer chromatography was performed using commercial thin-layer chromatography plates (Silica gel G 60, Merck). The eluant mixture was chloroform/methanol (95:5, v/v).
Microanalyses for carbon, hydrogen and nitrogen were carried out by Chemical & MicroAnalytical Services (Belmont, VIC, Australia). All other reagents and solvents were obtained from standard commercial sources and were used as received. CuII(atsm) [6, 31, 47], CuII(gtsm) [31, 48] H2atsm/a and H2atse/a were prepared by literature procedures [24–26].
Synthesis
Synthesis of H2 L 1
To a suspension of H2atsm/a (0.717 g, 2.74 mmol) in EtOH (20 mL) was added 1-pyrenecarboxaldehyde (0.632 g, 2.74 mmol) and the mixture was heated at reflux under an atmosphere of N2 for 4 h. A yellow solid was collected by filtration and washed with EtOH and Et2O (0.717 g, 79%). 1H NMR (d 6-DMSO) (400 MHz): δ 2.28, 3H, s, CH3; 2.35, 3H, s, CH3; 3.03, 3H, d, 3 J HH = 4.8 Hz, NH–CH 3; 8.11–8.36, 9H, m, ArH; 8.45, 1H, m, NH; 8.57, 1H, m, HC=N; 10.26, 1H, s, NH; 10.81, 1H, s, NH. 13C NMR (400 MHz): δ 19.3, CH3C=N; 31.9, NH–CH3; 124.4, 124.8, 125.9, 126.7, 126.9, 127.3, 128.1, 129.1, 129.4, 129.5, 129.7, 131.6, Ar; 133.5, CHC=N; 147.3, C=N; 179.2, C=S. Mass spectrometry: (positive ion) m/z 474 [L 1H2 + H+]+. HPLC: R T = 19.4 min.
Synthesis of H2 L 2
Following the same procedure employed for H2 L 1, 1-pyrenecarboxaldehyde (0.214 g, 0.931 mmol) and H2atse/a (0.256 g, 0.931 mmol) were used to prepare H2 L 2. The brown solid was washed with EtOH and Et2O (0.391 g, 87%). 1H NMR (d 6-DMSO) (400 MHz): δ 1.12, 3H, t, 3 J HH = 7.2 Hz, CH3; 2.26, 3H, s, CH3C=N; 2.33, 3H, s, CH3C=N; 3.58, 2H, m, NH–CH 2; 8.09–8.34, 9H, m, ArH; 8.47, 1H, m, NH; 8.54, 1H, d, 3 J HH = 8.4 Hz, ArH; 10.20, 1H, s, NH; 10.79, 1H, s, NH. Mass spectrometry: (negative ion) m/z 486 [L4H2–H+]−. HPLC: R T = 21.9 min.
Synthesis of ZnII L 1
To a suspension of H2 L 1 (0.317 g, 0.669 mmol) in EtOH (20 mL) was added zinc acetate dihydrate (0.147 g, 0.669 mmol) and the mixture was heated at reflux under an atmosphere of N2 for 4 h. An orange-red solid was collected by filtration and washed with EtOH and Et2O (0.291 g, 81%) (found: C, 53.7; H, 3.91; N, 18.3; calcd for ZnC24H23N7S2: C, 53.9; H, 3.96; N, 18.3). 1H NMR (d 6-DMSO) (400 MHz): δ 2.24, 3H, s, CH3; 2.34, 3H, s, CH3; 2.83, 3H, s, NH-CH 3; 8.08, 1H, t, 3 J HH = 8.0 Hz, ArH; 8.18, 1H, d, 3 J HH = 8.8 Hz, ArH; 8.19, 1H, d, 3 J HH = 8.8 Hz, ArH; 8.30, 4H, m, ArH; 8.41, 1H, d, 3 J HH = 8.4 Hz, ArH; 9.02, 1H, d, 3 J HH = 9.6 Hz, ArH; 9.08, 1H, s, HC=N; 11.53, 1H, s, NH. 13C NMR (400 MHz): δ 13.82, 14.15, CH3C=N; 18.52, NH–CH3; 123.4, 125.2, 125.4, 125.8, 126.5, 127.4, 127.8, 128.4, ArH; 123.9, 124.3, 127.7, 128.0, 130.3, 130.9, 131.0, Ar; 140.8, HC=N; 148.9, CH3 C=N. Mass spectrometry: (positive ion) m/z 536 {[ZnL1] + H+}+. HPLC: R T = 20.9 min.
Synthesis of ZnII L 2
Following the same procedure employed for ZnII L 1, zinc acetate dihydrate (0.149 g, 0.679 mmol) and H2 L 2 (0.331 g, 0.679 mmol) were used to prepare ZnII L 2. The dark-red solid was washed with EtOH and Et2O (0.338 g, 90%) (found: C, 54.6; H, 4.17; N, 17.8; calcd for ZnC25H23N7S2: C, 54.5; H, 4.21; N, 17.8). 1H NMR (d 6-DMSO) (400 MHz): δ 1.10, 3H, t, 3 J HH = 7.2 Hz, CH3; 2.21, 3H, s, CH3C=N; 2.31, 3H, s, CH3C=N; 3.40, 2H, m, NH–CH 2; 8.06, 1H, t, 3 J HH = 8.0 Hz, ArH; 8.14, 1H, d, 3 J HH = 9.2 Hz, ArH; 8.18, 1H, d, 3 J HH = 8.8 Hz, ArH; 8.29, 4H, m, ArH; 8.38, 1H, d, 3 J HH = 8.4 Hz, ArH; 9.01, 1H, d, 3 J HH = 9.6 Hz, ArH; 9.06, 1H, s, HC=N; 11.53, 1H, s, NH. Mass spectrometry: (positive ion) m/z 550 {[ZnL2] + H+}+. HPLC: R T = 21.9 min.
Synthesis of CuII L 1
To a suspension of H2 L 1 (0.354 g, 0.746 mmol) in EtOH (20 mL) was added copper acetate monohydrate (0.149 g, 0.746 mmol) and the mixture was heated at reflux under an atmosphere of N2 for 4 h. A dark-brown solid was collected by filtration and washed with EtOH and Et2O (0.306 g, 77%). An alternative method was by the transmetallation of ZnII L 1. To a suspension of ZnII L 1 (0.130 g, 0.274 mmol) in DMF (5 mL) was added copper acetate monohydrate (0.109 g, 0.549 mmol) and the mixture was stirred at room temperature under an atmosphere of N2 for 30 min. A dark-brown solid was precipitated with water and collected by filtration. The solid was washed with EtOH and Et2O (0.118 g, 80%). Mass spectrometry: (positive ion) m/z 535 {[CuII L 1] + H+}+. HPLC: R T = 20.9 min.
Synthesis of CuII L 2
Following the same procedure employed for CuII L 1, copper acetate monohydrate (0.0849 g, 0.425 mmol) and H2 L 2 (0.207 g, 0.425 mmol) were used to prepare CuII L 2. The dark-brown solid was washed with EtOH and Et2O (0.131 g, 56%) (found: C, 54.9; H, 4.19; N, 18.0; calcd for CuC25H23N7S2: C, 54.7; H, 4.22; N, 17.9). HPLC: R T = 19.4 min.
Stability of CuII L 1 in 20% DMSO solution and in the presence of glutathione, l-cysteine and l-histidine
Solutions consisting of (1) 10 μM CuII L 1 in 20% v/v DMSO/PB solution, (2) 10 μM CuII L 1 and 100 μM glutathione in 20% v/v DMSO/PB solution and (3) 10 μM CuII L 1 and 100 μM l-cysteine and l-histidine in 20% v/v DMSO/PB solution were prepared and kept in a 37 °C water bath. Aliquots (150 μL) of the solutions were removed at 0, 1, 2, 3 and 4 h for HPLC measurements.
Radiolabelling with 64Cu
64CuCl2 (1.80 GBq/mL, pH 1) was purchased from ANSTO Radiopharmaceuticals and Industrials (Lucas Heights, NSW, Australia). The radionuclidic purity at calibration {(64Cu)/(67Cu)} was more than 99.9% and the radiochemical purity as CuII was more than 99.9%. The chemical purities of copper, zinc and iron were 2.1, 8.1 and less than 1.0 μg/mL, respectively. A solution consisting of aqueous sodium acetate (0.1 M, 90 μL), distilled water (390 μL) and ZnIIL1 in DMSO (1 mg/mL, 10 μL) was prepared. 64CuL1 (1.8 MBq/μg of ZnIIL1) was prepared by reacting aqueous 64CuCl2 (pH 1, 10 μL, 18 MBq) with the previously mentioned solution at room temperature. The synthesis was essentially complete after 5 min and 100 μL of the reaction solution was taken for analysis by reverse-phase radio-HPLC. HPLC analysis was performed using a Shimadzu LC20AT HPLC instrument with a 150 mm × 4.6 mm Waters Cosmosil/Cosmogel C18 column eluted at 1 mL/min for 25 min with a solvent gradient of water/trifluoroacetic acid (0.1%) and acetonitrile/trifluoroacetic acid (0.1%): initially 0%/100%, then 100%/0% at 22 min. Both UV detection (λ = 254 nm) and scintillation detection (NaI) were used in series. The radiochemical yield was more than 95% based on HPLC analysis (‘free’ 64Cu has RT = 1.9 min).
Cell culture
HeLa cells were grown continuously and passaged at regular intervals. Cells were used once approximately 80% confluency had been reached.
Cell uptake and washout studies
The compounds were prepared at 10 mM in DMSO (culture-grade DMSO available in the cell culture laboratory). Twenty-five micromolar mixtures of the compound and the medium were generated by adding 2.5 μL of the compound solution to 1 mL of medium (serum-free OptiMem). The medium was removed from the cells and was replaced by the medium/compound mix. Each compound was tested in triplicate. The incubation temperature was 37 °C. The cells with the compound were incubated, treated and harvested in four different ways; the cells were harvested after 30-min incubation; the medium/compound mix was removed and replaced with fresh medium after 30 min and cells were harvested after 120 min; the medium/compound mix was removed and replaced with fresh medium after 30 and 60 min and cells were harvested after 120 min; the cells were harvested after 120 min. DMSO (25 μL) was used as a vehicle control whereby cells were treated and incubated for 30 and 120 min. Scraping the cells into the medium and transferring the medium/cell into an Eppendorf tube harvested the cells. The tubes were centrifuged for 2–3 min at 14,000 rpm and the medium was removed and discarded. PB solution (0.5 mL) (KCl 0.2 g/L, KH2PO4 0.2 g/L, Na2HPO4 1.15 g/L, NaCl 8 g/L) was added to wash the cells and they were resuspended. The tubes were recentrifuged for 2–3 min at 14,000 rpm and the PB supernatant was discarded. The cell pellets were stored at −20 °C. CuII(atsm), CuII(gtsm) and DMSO cell uptake were used as controls. Multielement analysis was performed using a Varian UltraMass ICP-MS instrument. The instrument was calibrated using a blank and 10, 50 and 100 ppb of a certified multielement ICP-MS standard solution (ICP-MS-CAl2-1, Accustandard) for manganese, iron, copper and zinc in 1% nitric acid. Metal levels were determined in cell pellets by ICP-MS as described previously and converted to the fold increase in the metal compared with untreated controls [40].
Hypoxic cell studies
Prior to treatment with CuII L 1, HeLa cells in a 24-well-plate format were placed in the BD GasPak™ EZ gas generating chamber with two activated gas-generating sachets for 6 h (BD Biosciences, North Ryde, Australia). These sachets are designed to remove oxygen and generate carbon dioxide, and contain a mixture of inorganic carbonate, activated carbon, ascorbic acid and water. The BD GasPak™ EZ gas generating chamber with two activated gas-generating sachets is capable of generating atmospheres of less than 0.4% O2 [45, 46]. Low oxygen was confirmed in each assay as per the manufacturer’s instructions. Following this, the plate was briefly removed from the chamber, treated with CuII L 1, then immediately placed back into the chamber with two fresh gas-generating sachets for the duration of the treatment.
Fluorescence studies
Human carcinoma HeLa cells were grown on poly(l-lysine)-coated cover slips in a 24-well plate and incubated at 37 °C (95% air and 5% CO2). When the cells had reached approximately 80% confluency, they were treated with 50 μM CuL 1, 0.5 μM nuclear dye To-Pro-3 iodide (Molecular Probes/Invitrogen, Melbourne, Australia; λex = 633 nm, λem = 642–670 nm) and either 75 nM LR (Molecular Probes/Invitrogen, Melbourne, Australia; λex = 543 nm, λem = 572–610 nm) or 6.76 μM acridine orange (Sigma, Sydney, Australia; λex = 488 nm, λem = 600–645 nm). Cell treatments were all for 30 min, except for LR, which was incubated with cells for up to 2 h. The cells were then rinsed twice with 1× PB solution and fixed with 4% (w/v) paraformaldehyde for 20 min at room temperature. Following fixation, the cells were rinsed twice with 1× PB solution and mounted onto glass slides using DAKO® fluorescence mounting medium. Images of the cells were collected using an Olympus FV1000 IV81 confocal microscope.
Abbreviations
- H2atse/a:
-
Diacetyl bis(N 4-ethyl-N 4′-amino-thiosemicarbazone)
- H2atsm:
-
Diacetyl bis(N 4-methylthiosemacarbazone)
- H2atsm/a:
-
Diacetyl bis(N 4-methyl-N4′-amino-thiosemicarbazone)
- CuII(atsm):
-
Diacetyl bis(N 4-methylthiosemicarbazonato)copper(II)
- CuII(gtsm):
-
Glyoxalbis(N 4-methylthiosemicarbazonato)copper(II)
- DMF:
-
Dimethylformamide
- d6-DMSO:
-
Deuterated dimethyl sulphoxide
- DMSO:
-
Dimethyl sulphoxide
- EPR:
-
Electron paramagnetic resonance
- ES-MS:
-
Electrospray mass spectrometry
- ICP-MS:
-
Inductively coupled plasma mass spectrometry
- LR:
-
LysoTracker Red
- PB:
-
Phosphate buffer
- SCE:
-
Saturated calomel electrode
References
Blower PJ, Lewis JS, Zweit J (1996) Nucl Med Biol 23:957
Smith SV (2004) J Inorg Biochem 98:1874
Denny WA (2004) Aust J Chem 57:821–828
Fujibayashi Y, Taniuchi H, Yonekura Y, Ohtani H, Konishi J, Yokoyama A (1997) J Nucl Med 38:1155–1160
Dearling JLJ, Lewis JS, Mullen GED, Rae MT, Zweit J, Blower PJ (1998) Eur J Nucl Med 25:788–792
Dearling JLJ, Lewis JS, Mullen GD, Welch MJ, Blower PJ (2002) J Biol Inorg Chem 7:249–259
Fujibayashi Y, Taniuchi H, Wada K, Yonekura Y, Konishi J, Yokoyama A (1995) Ann Nucl Med 9:1–5
Obata A, Yoshimi E, Waki A, Lewis JS, Oyama N, Welch MJ, Saji H, Yonekura Y, Fujibayashi Y (2001) Ann Nucl Med 15:499
Lewis JS, Laforest R, Buettner TL, Song S-K, Fujibayahsi Y, Connet JM, Welch MJ (2001) Proc Natl Acad Sci 98:1206
Lewis JS, Sharp TL, Laforest R, Fujibayahsi Y, Welch MJ (2001) J Nucl Med 42:655
Cowley AR, Dilworth JR, Donnelly PS, Labisbal E, Sousa A (2002) J Am Chem Soc 124:5270–5271
Holland JP, Green JC, Dilworth JR (2006) Dalton Trans 783–794
Vavere AL, Lewis JS (2007) Dalton Trans 4893–4902
Dearling JLJ, Lewis JS, McCarthy DW, Welch MJ, Blower PJ (1998) J Chem Soc Chem Commun 2531
Xiao Z, Donnelly PS, Zimmermann M, Wedd AG (2008) Inorg Chem 47:4338–4347
Donnelly PS, Caragounis A, Du T, Laughton KM, Volitakis I, Cherny RA, Sharples RA, Hill AF, Li Q-X, Masters CL, Barnham KJ, White AR (2008) J Biol Chem 283:4568–4577
Maurer RI, Blower PJ, Dilworth JR, Reynolds CA, Zheng Y, Mullen GED (2002) J Med Chem 45:1420
Burgman P, O’Donoghue JA, Lewis JS, Welch MJ, Humm JL, Ling CC (2005) Nucl Med Biol 32:623–630
Obata A, Kasamatsu S, Lewis JS, Furukawa T, Takamatsu S, Toyohara J, Asai T, Welch MJ, Adams SG, Saji H, Yonekura Y, Fujibayashi Y (2005) Nucl Med Biol 32:21–28
Cowley AR, Davis J, Dilworth JR, Donnelly PS, Dobson R, Nightingale A, Peach JM, Shore B, Kerr D, Seymour L (2005) Chem Commun 845–847
Haefliger P, Agorastos N, Renard A, Giambonini-Brugnoli G, Marty C, Alberto R (2005) Bioconjug Chem 16:582–587
Haefliger P, Agorastos N, Spingler B, Georgiev O, Viola G, Alberto R (2005) Chembiochem 6:414–421
Pouget J-P, Mather SJ (2001) Eur J Nucl Med 28:541–561
Christlieb M, Cowley AR, Dilworth JR, Donnelly PS, Paterson BM, Struthers HSR, White JM (2007) Dalton Trans 327–331
Holland JP, Aigbirhio FI, Betts HM, Bonnitcha PD, Burke P, Christlieb M, Churchill GC, Cowley AR, Dilworth JR, Donnelly PS, Green JC, Peach JM, Vasudevan SR, Warren JE (2007) Inorg Chem 46:465–485
Christlieb M, Struthers HSR, Bonnitcha PD, Cowley AR, Dilworth JR (2007) Dalton Trans 5043–5054
Petering DH (1974) Biochem Pharmacol 23:567–576
Bushnell GW, Tsang AYM (1979) Can J Chem 57:603
John E, Fanwick PE, McKenzie AT, Stowell JG, Green MA (1989) Nucl Med Biol 16:791–797
Castiñeiras A, Bermejo E, West DX, El-Sawaf AK, Swearingen JK (1998) Polyhedron 17:2751–2757
Blower PJ, Castle TC, Cowley AR, Dilworth JR, Donnelly PS, Labisbal E, Sowrey FE, Teat SJ, Went MJ (2003) Dalton Trans 4416–4425
Alsop L, Cowley AR, Dilworth JR, Donnelly PS, Peach JM, Rider JT (2005) Inorg Chim Acta 358:2770–2780
Warren LE, Horner SM, Hatfield WE (1972) J Am Chem Soc 94:6392–6396
Rodriguez-Arguelles MC, Battaglia LP, Ferrari MB, Fava GG, Pelizzi C, Pelosi G (1995) J Chem Soc, Dalton Trans 2297–2303
Castineiras A, Bermejo E, West DX, Ackerman LJ, Valdes-Martinez J, Hernandez-Ortega S (1999) Polyhedron 18:1463–1469
Matkovich KM, Thorne LM, Wolf MO, Pace TCS, Bohne C, Patrick BO (2006) Inorg Chem 45:4610–4618
Winnik FM (1993) Chem Rev 93:587–614
Bonnitcha PD, Vavere AL, Lewis JS, Dilworth JR (2008) J Med Chem 51:2985–2991
White AR, Du T, Laughton KM, Volitakis I, Sharples RA, Xilinas ME, Hoke DE, Holsinger RMD, Evin G, Cherny RA, Hill AF, Barnham KJ, Li Q-X, Bush AI, Masters CL (2006) J Biol Chem 281:17670–17680
Caragounis A, Du T, Filiz G, Laughton KM, Volitakis I, Sharples RA, Cherny RA, Masters CL, Drew SC, Hill AF, Li Q-X, Crouch PJ, Barnham KJ, White AR (2007) Biochem J 407:435–450
Jansen BAJ, Wielaard P, Kalayda GV, Ferrari M, Molenaar C, Tanke HJ, Brouwer J, Reedijk J (2004) J Biol Inorg Chem 9:403–413
New EJ, Duan R, Zhang JZ, Hambley TW (2009) Dalton Trans 3092–3101
Schweichel JU, Merker HJ (1973) Teratology 7:253–266
Mizushima N (2004) Int J Biochem Cell Biol, Cell Biol 36:2491–2502
Sapna S, Shivakumar K (2007) Mol Cell Biochem 303:259–262
Kendler A, Dawson G (1990) J Biol Chem 265:12259–12266
Gingras BA, Suprunchuk T, Bayley CH (1962) Can J Chem 40:1053
Beraldo H, Boyd LP, West DX (1998) Transition Met Chem 23:67–71
Acknowledgments
This research was funded by the Australian Research Council, the National Health and Medical Research Council of Australia and the Australian Institute of Nuclear Science and Engineering. We thank James Camakaris for our ongoing collaboration with provision of 64Cu, Irene Volitakis and Robert A. Cherny for ICP-MS analysis, Giuseppe D. Ciccotosto for assistance with preliminary confocal microscopy measurements and Kenneth Ghiggino for advice on fluorescence measurements. B.M.P. acknowledges the support of an Australian Postgraduate Award and S.L. acknowledges the support of a Science Faculty Scholarship (University of Melbourne).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Lim, S., Price, K.A., Chong, SF. et al. Copper and zinc bis(thiosemicarbazonato) complexes with a fluorescent tag: synthesis, radiolabelling with copper-64, cell uptake and fluorescence studies. J Biol Inorg Chem 15, 225–235 (2010). https://doi.org/10.1007/s00775-009-0587-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-009-0587-4