
Abstract
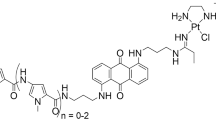
A series of N,N′-bis(aminoalkyl)-1,4-diaminoanthraquinones (aminoalkyl=2-aminoethyl, 3-aminoprop-1-yl and 4-aminobut-1-yl) was functionalized with trans-platinum DNA-binding moieties. Cytotoxicity testing in A2780 human ovarian carcinoma cells revealed high anticancer activity of the formed cationic dinuclear platinum complexes. The cationic dinuclear platinum complexes with the shortest aminoalkyl chain were shown to be the most active, which agrees with the structure–activity relationship found for the corresponding free ligands without platinum. The N,N′-bis(aminoalkyl)-1,4-diaminoanthraquinones partly circumvent cisplatin resistance, whereas their dinuclear platinum complexes were found susceptible to the resistance mechanisms in A2780cisR. The platinum complexes have resistance factors comparable to the control dinuclear complex BBR3005 [{trans-PtCl(NH3)2}2{μ-(NH2(CH2)6NH2)}](NO3)2. The 1,4-diaminoanthraquinone moiety is fluorescent, and thus the cellular processing of the compounds could be monitored by time-lapse digital fluorescence microscopy. The intercalators without platinum were shown to enter the cells within minutes. The platinum complexes enter the cells more slowly. Most likely, the positive charges of the platinum complexes hamper the diffusion through the membrane. Interestingly, the platinum complexes are processed differently than the platinum-free compounds by the cells. After 24 hours the fluorescent platinum complexes are encapsulated in large vesicles in the cytosol. Co-localization of the anthraquinone fluorescence with Lysotracker Green DND-26 shows that these vesicles are acidic compartments, probably lysosomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dinuclear and polynuclear cationic platinum complexes, that are structurally very different from established platinum drugs such as cisplatin and carboplatin, are a promising class of new platinum anticancer drugs [1, 2, 3, 4, 5]. Many compounds from this group of complexes show antitumor activity [6, 7, 8, 9, 10, 11] and circumvent cisplatin resistance [6, 7, 8, 9, 12, 13, 14, 15]. Synthetic variations in coordinating ligands, as well as the linkers that connect the platinum nuclei, have led to new, active platinum complexes. Dinuclear platinum complexes based on the polyamines spermine and spermidine [16], as well as the trinuclear complex BBR3464 [6], have linkers that bear additional positive charges, resulting in a high affinity for the presumed cellular target of platinum antitumor drugs, i.e. the nuclear DNA [17]. Moreover, these complexes show very high cytotoxic activities [6, 16]. In the current paper, a series of dinuclear platinum complexes is presented that accomplish an additional interaction with DNA through an intercalating moiety which is incorporated in the linker.
Bis(aminoalkyl)diaminoanthraquinones with various lengths of the aminoalkyl group (AQ2, AQ3 and AQ4 in Scheme 1) were selected as linkers for the dinuclear platinum complexes PAQ2, PAQ3 and PAQ4. These intercalators are known for their intrinsic cytotoxic activity [18, 19], and bear free primary amines available for coordination to platinum(II). In earlier studies it was shown that the replacement of the amines of cisplatin-type platinum drugs with anthraquinone [20, 21, 22, 23, 24] or acridine [25, 26, 27, 28, 29, 30] intercalators may yield anticancer-active platinum compounds. Platinum–intercalator complexes were shown to bind differently to DNA compared to cisplatin [28, 31, 32], which may help to circumvent cisplatin resistance [33]. An interesting aspect of diaminoanthraquinones is that these molecules are fluorescent, and may thus be studied by techniques such as fluorescence microscopy [34]. Recently, it was shown that cisplatin derivatives labelled with fluorescent probes provide new insights into the cellular processing of platinum anticancer drugs [35]. Fluorescent probes reveal how platinum complexes enter the cell, where they are located inside the cell and how the drugs are excreted. As the uptake mechanisms and cellular transport of polynuclear cationic platinum complexes remain largely unknown [8, 9], a fluorescent-labelled dinuclear complex may improve our understanding of this new type of platinum anticancer compounds.

Synthesis of the dinuclear platinum complexes with N,N′-bis(aminoalkyl)-1,4-diaminoanthraquinones as linkers
In the present paper, a series of dinuclear cationic platinum complexes based on N,N′-bis(aminoalkyl)-1,4-diaminoanthraquinones as fluorescent linking ligands is described. The N,N′-bis(aminoalkyl)-1,4-diaminoanthraquinones are suitable for the standard functionalization synthesis of alkylenediamines with [trans-PtCl(DMF)(NH3)2] (Scheme 1) [15, 36, 37]. Both free intercalators and platinum complexes were tested for cytotoxicity in A2780 human ovarian carcinoma cells, and the cisplatin-resistant derivative A2780cisR. The anthraquinone moiety functions as a fluorescent probe, and thus the cellular processing of the compounds could be monitored by time-lapse digital fluorescence microscopy.
Materials and methods
General
Chemicals
1,4-Difluoroanthraquinone, silver nitrate and the diaminoalkanes (1,2-diaminoethane, 1,3-diaminopropane and 1,4-diaminobutane) were obtained from Aldrich. trans-PtCl2(NH3)2 was prepared according to a literature procedure [38]. The dinuclear platinum complex BBR3005 [{trans-PtCl(NH3)2}2{μ-(NH2(CH2)6NH2)}](NO3)2 was synthesized as described previously [36]; mitoxantrone was purchased from Sigma.
Analysis
NMR spectra were recorded on a Bruker DPX 300 spectrometer with a 5 mm multi-nucleus probe. The temperature was kept constant at 25 °C using a variable temperature unit. 1H NMR spectra were measured in D2O using TMS as an external reference at δ=0 ppm. 195Pt spectra were calibrated with respect to external K2PtCl4 at δ=−1614 ppm. The solubility of some complexes in D2O was too low for achieving a sufficient 195Pt signal-to-noise ratio within a reasonable recording time, and the 195Pt spectra of these complexes were therefore recorded in (undeuterated) DMF. C, H and N analyses were performed on a Perkin-Elmer 240B microanalyzer.
Synthesis
N,N′-Bis(2-aminoethyl)-1,4-diaminoanthracene-9,10-dione
To a solution of 1,4-difluoroanthraquinone (240 mg, 1 mmol) in pyridine (5 mL) a solution of 1,2-diaminoethane (240 mg, 4 mmol) in pyridine (5 mL) was added. The solution was stirred at room temperature for 79 h. Pentane (10 mL) was added to the mixture and the blue precipitate was collected by filtration. The crude product was dissolved in chloroform, and insoluble material was filtered off. Pentane (200 mL) was added to the filtrate, and the solution was allowed to stand overnight at 4 °C. The blue AQ2 product was filtered off, washed with cold diethyl ether and dried in a vacuum desiccator over KOH; yield: 58%. 1H NMR (CDCl3): δ (ppm)=10.90 (b, 2NH), 8.35 (dd, 2H), 7.70 (dd, 2H), 7.25 (s, 2H), 3.51 (q, 4H), 3.05 (t, 4H)
N,N′-Bis(3-aminoprop-1-yl)-1,4-diaminoanthracene-9,10-dione
1,4-Difluoroanthraquinone (315 mg, 1.3 mmol) was added to 1,3-diaminopropane (25 mL, 300 mmol). The solution was stirred at room temperature for 5 days. The excess 1,3-diaminopropane was evaporated under reduced pressure. To the blue residue, chloroform (50 mL) was added. The solution was heated and insoluble material was filtered off. Next, pentane (400 mL) was added to the filtrate. The solution was allowed to stand overnight at 4 °C. The blue AQ3 product was filtered off, washed with cold diethyl ether and dried in a vacuum desiccator over KOH; yield: 66%. 1H NMR (CDCl3): δ (ppm)=10.82 (b, 2NH), 8.34 (dd, 2H), 7.69 (dd, 2H), 7.28 (s, 2H), 3.51 (q, 4H), 2.93 (t, 4H), 1.91 (m, 4H).
N,N′-Bis(4-aminobut-1-yl)-1,4-diaminoanthracene-9,10-dione
1,4-Diaminobutane (22 mg, 249 mmol) was heated at 35 °C, and 1,4-difluoroanthraquinone (253 mg, 1.04 mmol) was added to yield a solution that turned blue almost immediately. The solution was stirred at 35 °C for 5 days. Subsequently, chloroform (50 mL) was added, and the resulting solution was washed repeatedly (5–10 times) with small amounts of water (10 mL), to remove excess. Then chloroform was evaporated under reduced pressure; the blue AQ4 product remained, which was dried in a vacuum desiccator over KOH; yield: 80%. 1H NMR (CDCl3): δ (ppm)=10.83 (b, 2NH), 8.33 (dd, 2H), 7.69 (dd, 2H), 7.25 (s, 2H), 3.44 (q, 4H), 2.78 (t, 4H), 1.81 (m, 4H), 1.65 (m, 4H)
[{trans-PtCl(NH3)2}2(μ-AQ2)](NO3)2
To a solution of trans-[PtCl2(NH3)2] (172 mg, 0.57 mmol) in DMF (5 mL) a solution of AgNO3 (98 mg, 0.58 mmol) in DMF (5 mL) was added. The solution was stirred in the dark overnight at room temperature. The precipitated AgCl was filtered off. A yellow filtrate remained. A solution of AQ2 (92.9 mg, 0.29 mmol) in DMF (5 mL) was added, and the resulting solution was stirred overnight in the dark. After that, diethyl ether (400 mL) was added and a blue precipitate appeared. The solution was allowed to stand overnight at 4 °C. The precipitate was filtered off the red solution and washed with diethyl ether. Methanol (75 mL) was added to the blue compound and this suspension was stirred for 2.5 h, to remove DMF. The solid was filtered off and washed with ethanol and diethyl ether, and dried under vacuum over KOH. The complex was purified by recrystallization from boiling methanol (60 mL). The PAQ2 complex was filtered off, washed with ethanol and diethyl ether, and dried under vacuum; yield: 22%. 1H NMR (D2O): δ (ppm)=8.25 (m, 2H), 7.82 (m, 2H), 7.53 (s, 2H), 3.7 (m, 4H), 2.9 (m, 4H); 195Pt NMR (D2O): δ (ppm)=−2394; anal. for C18H32Cl2N10O8Pt2: % found (calculated): C 22.92 (22.13), H 3.36 (3.30), N 14.15 (14.35).
[{trans-PtCl(NH3)2}2(μ-AQ3)](NO3)2
This dinuclear complex (PAQ3) was obtained following the same procedure as for PAQ2; yield: 39%. 1H NMR (D2O): δ (ppm)=8.02 (m, 2H), 7.40 (m, 2H), 7.07 (s, 2H), 3.44 (m, 4H), 2.83 (m, 4H), 2.09 (m, 4H); 195Pt NMR (DMF): δ(ppm)=−2403; anal. for C20H36Cl2N10O8Pt2: % found (calculated): C 23.80 (23.89), H 3.67 (3.61), N 13.77 (13.93).
[{trans-PtCl(NH3)2}2(μ-AQ4)](NO3)2
This dinuclear complex (PAQ4) was obtained following the same procedure as for PAQ2; yield: 60%. 1H NMR (D2O): δ (ppm)=8.10 (m, 2H), 7.77 (m, 2H), 7.21 (s, 2H), 3.39 (m, 4H), 2.72 (m, 4H), 1.80 (m, 8H); 195Pt NMR (DMF): δ(ppm)=−2401; anal. for C20H36Cl2N10O8Pt2: % found (calculated): C 24.87 (25.56), H 3.91 (3.90), N 13.63 (13.55).
Growth inhibition assays in A2780 and A2780cisR cells
A2780 and A2780cisR (cisplatin resistant) human ovarian cell lines originate from a gift of Dr. J.M. Perez (Universidad Autónoma de Madrid, Spain). The cell lines were grown as monolayers in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (Gibco, Paisley, UK), penicillin (100 units/mL; Duchefa, Netherlands) and streptomycin (100 µg/mL; Duchefa, Netherlands) in a humidified 6% CO2/94% air atmosphere.
Growth inhibition by the compounds was determined using an MTT-based assay [39]. After trypsinization, cells were divided into 96-well plates at concentrations of 3–5×103 cells/well in 100 μL growth medium. The cells were allowed to attach overnight. After 24 h, fresh stock solutions (1 mg/mL) of the compounds in Millipore water were diluted in the medium. For the compounds AQ2, AQ3 and AQ4, the stock solutions were slightly acidified (pH 3–4) with hydrochloric acid for improved solubility. Five subsequent dilutions of each complex were added to the cells in quadruplicate, yielding a total volume of 200 μL in each well. After 72 h of incubation, 50 μL of a 5 mg/mL MTT solution in phosphate buffered saline (PBS) was added to each well, and allowed to develop in the incubator (usually 60 min). After this, the medium was discarded and 100 μL of DMSO was added to each well, yielding purple solutions. The optical density was measured at 590 nm using a Biorad 550 microplate reader. The IC50 values (drug concentration that inhibits cell growth for 50% with respect to control) were determined graphically using the Graphpad Prism analysis software package.
Nuclear DNA binding studies
For the experiment, A2780 cells were grown in 6-cm plates with 4 mL of DMEM medium until a concentration of approximately 1×107 cells was reached. The cells were washed with serum-free DMEM medium, and the platinum complexes, freshly dissolved in serum-free DMEM medium at a concentration of 200 μM, were added to the cells. Each experiment was performed in duplicate. After 1 h of incubation (37 °C, 6% CO2, dark) the medium was removed and the cells were washed twice with PBS. Then the cells were trypsinized and centrifuged at 1000×g for 10 min. At this point, the cell pellets may be stored at −20 °C. Nuclear DNA was isolated from the cells using the following procedure. The cell pellets were suspended in 300 μL of the buffer (10 mM Tris, pH 8, 150 mM NaCl, 10 mM EDTA). Then 5 μL of proteinase K solution was added and mixed well with the cells. Finally, 50 μL of 10% sodium dodecyl sulfate was added, and the resulting mixture was heated at 50–55 °C for 1.5 h. The solutions were transferred to 2-mL Eppendorf Phaselock tubes and extracted with 300 μL of chloroform/pentyl alcohol (24:1). DNA was precipitated by adding 600 μL of isopropyl alcohol to the water layer. The DNA pellet was washed once with cold 70% ethanol and then dissolved in 1 mL of Milli-Q water. The DNA concentration was determined by measuring the UV absorption at 260 nm. From the absorption values, the concentration of the base pairs was calculated using the average molar extinction coefficient per base pair of ε260=16,800 M−1 cm−1. The platinum concentration was measured by FAAS. From the platinum concentration, the actual drug concentration was derived. Combining FAAS and UV results, the drug molecules per base pair ratio (r b) was calculated.
Digital fluorescence microscopy
Fluorescence microscopy experiments were performed on an Axiovert 135 TV (Zeiss, Jena, Germany) inverted microscope, equipped with a 100 W mercury arc lamp for fluorescence excitation and bright-field illumination for phase contrast images. A Texas Red filter set (Chroma Technology, Brattleboro, Vt., USA) was used to detect the 1,4-diaminoanthraquinone fluorescence, and consisted of a hq 560/55 band-pass excitation filter, a hq 645/75 band-pass emitter filter and a Q595 long-pass beamsplitter. The temperature of the culture medium was controlled between 36–37 °C by a Bioptechs (Butler, Pa., USA) objective heater and a heated ring surrounding the culture chamber. Observations were made through a Neofluor 40× NA 1.30 oil-immersion objective (Zeiss, Jena, Germany). Digital images were taken with a cooled CCD camera (Photometrix PLX, Tucson, Ariz., USA).
Fluorescence microscopy uptake experiments
Fluorescence microscopy experiments were performed similarly to the work previously described by some of us [35]. For the living cell observations, A2780 cells were grown to 30–50% confluence in DMEM without phenol red (Gibco) in 35 mm culture dishes with a glass cover slip incorporated in the bottom (Mattek, Ashland, Mass., USA), which is directly in contact with the heated oil-immersion objective during observations. Before the uptake experiments, the cells were washed with PBS. Subsequently, the complexes were added to the cells at a final concentration of 3 μM in serum-free medium. Phase contrast and corresponding fluorescence images were taken at several time points during the incubation.
Uptake of the drugs by A2780 human ovarian carcinoma cells was studied over time. These cells showed negligible interfering autofluorescence at the wavelengths employed for the anthraquinone compounds. Therefore, in the quantification of fluorescence emission, the autofluorescence was not taken into account. Immediately after addition of the drugs, images of the cells were taken. The fluorescence inside the cell was quantified digitally by integration of the total fluorescence in the cell area as recorded on the digital fluorescence images. Quantification was done with in-house image analysis software. The exposure times were varied in the range between 0.1–1 s, because a large variation in intensities was observed. Exposure times were adjusted to a fluorescence intensity level significantly above background emission, without exceeding the signal overflow value. For quantification of emission, the intensities were normalized to a 1 s exposure time. The mean fluorescence emission intensity per pixel, F mean, was calculated by:
where F i is the mean intracellular emission intensity value per pixel (in arbitrary units of fluorescence intensity, alufi) measured in an area limited by the outer cell membrane, selected by drawing the area by hand; F e is the background emission per pixel measured in a cell-less area; and t exp is the exposure time in seconds. For each determination, at least 10 well-adhered cells were measured, from at least three different images taken from different areas in the cell culture dish.
Fluorescence excitation and emission spectra
Fluorescence excitation and emission spectra were recorded in Milli-Q water on a Perkin-Elmer LS50B luminescence spectrophotometer. Fluorescence emission spectra were measured from 600 to 900 nm, with an excitation wavelength of 580 nm. Excitation spectra were recorded at 700 nm, with excitations ranging from 400 to 680 nm.
For the quenching experiment, small amounts of a concentrated stock solution of pBR322 plasmid DNA (Sigma) were added to 10 μM solutions of the various fluorescent molecules. After each addition, the solution was mixed with a pipette, and immediately the fluorescence emission spectrum was recorded. The fluorescence intensities of the solutions were found to be stable for at least 15 min. The final added volume was less than 2% of the total volume.
Golgi staining
Staining of the Golgi complex was performed with 6-{[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]hexanoyl}sphingosine (NBD C6-ceramide) complexed to bovine serum albumin (BSA) (Molecular Probes, Leiden, The Netherlands). After 20 min of incubation with AQ2 (and after 2 h of incubation with PAQ2), the cells were washed with serum-free medium. Then NBD C6-ceramide was added to the culture medium to a final concentration of 250 nM. After 10 min of incubation, the cells were rinsed with a complete medium and observed live using digital fluorescence microscopy. AQ2 and PAQ2, emitting red light, and NBD C6-ceramide, emitting in green, were sequentially visualized and digitally recorded.
Staining of lysosomes
LysoTracker Green DND-26 (Molecular Probes, Leiden, The Netherlands) was used for staining of the large vesicles observed in the cytosol after 24 h of incubation with the platinum complexes PAQ2, PAQ3 and PAQ4. After 24 h of incubation with PAQ2, LysoTracker Green DND-26 was added to the culture medium to a final concentration of 50 nM. After 30 min of incubation, the cells were observed live using digital fluorescence microscopy. PAQ2, emitting in red, and LysoTracker Green DND-26, emitting green light, were sequentially visualized and digitally recorded.
Results and discussion
Synthesis
The synthesis of the N,N′-bis(aminoalkyl)-1,4-diaminoanthraquinones was performed according to the method described by Krapcho et al. [19, 40]. This method was modified for the diaminopropane (AQ3) and diaminobutane (AQ4) compounds; by using the appropriate diaminoalkane as a solvent while extending the reaction time, the use of pyridine could be avoided. Furthermore, this improved method appears to yield less side products, and sufficiently pure products were isolated without cumbersome purification steps such as column chromatography. Diaminopropane may be removed by evaporation under reduced pressure, while less volatile diaminoalkanes were removed from solutions in organic solvents such as chloroform by repeated extraction with water.
The ligands AQ2, AQ3 and AQ4 are compatible with the common synthetic procedure for alkylenediamine-linked dinuclear trans platinum complexes. Adding the diamine linker to a solution of trans-PtCl(DMF)(NH3)2 yielded the dinuclear platinum complexes PAQ2, PAQ3 and PAQ4. Yields after purification were 22, 39 and 60%, respectively.
Cytotoxicity and resistance profiles
The anthraquinones and their dinuclear platinum complexes were tested for cytotoxicity in the A2780 human ovarian cancer cell line and the cisplatin-resistant A2780cisR cell line. Cisplatin, the dinuclear platinum complex BBR3005 [{trans-PtCl(NH3)2}2{μ-(NH2(CH2)6NH2)}](NO3)2 and the dihydroxyanthraquinone drug mitoxantrone were included as references. Results are presented in Table 1.
Against A2780 ovarian cancer, the tested compounds possess considerable anti-cancer potential, as all IC50 values are in the lower micromolar range. Comparing with the reference complexes cisplatin and mitoxantrone, it is clear that mitoxantrone is active at much lower concentrations. The cytotoxicity of the anthraquinones is in the order AQ4<AQ3<AQ2<<mitoxantrone. This sequence is comparable to the results found earlier in literature in other cell lines [18, 19]. Apparently, the longer aminoalkyl substituents make the complexes less cytotoxic. The same cytotoxicity trend is observed in the dinuclear platinum series, i.e. PAQ4<PAQ3<PAQ2.
The cisplatin resistance in the A2780cisR cell line is known to be multifocal, with increased glutathione levels and increased DNA repair as the main identified mechanisms [41, 42, 43, 44]. Interestingly, the reference complexes BBR3005 and mitoxantrone also appear to be susceptible to the resistance mechanisms in A2780cisR, even though their DNA adducts are expected to differ from those of cisplatin. In an earlier study, it was found that BBR3005 circumvents the resistance mechanisms in L1210 murine leukemia [3, 15], but the mechanisms in A2780cisR appear to be more effective against the dinuclear platinum complexes. Within the series, the compounds with the shortest aminoalkyl chain are the most active. The values of the resistance factors show that the compounds respond differently in the two cell lines. Although the ligands AQ2, AQ3 and AQ4 partly overcome resistance in A2780cisR, the platinum complexes PAQ2 and PAQ3 are significantly affected, with resistance factors (RF) even higher than those of cisplatin and BBR3005. More detailed investigation has revealed that several independent mechanisms contribute to the cross-resistance of the PAQ complexes with cisplatin: deactivation of the complexes in cytosol by glutathione and their accumulation in acidic vesicles [45].
Fluorescence microscopy: uptake and localization
Recently, it was shown that fluorescence microscopy is a powerful tool to obtain insight into the cellular processing of fluorophore-labelled cisplatin derivatives [35]. For the compounds in this study, no specific labelling was required, as the 1,4-diaminoanthraquinone moiety shows sufficiently intense fluorescence for microscopic purposes [34]. In Fig. 1 the excitation spectrum and emission spectrum of the 1,4-diaminoanthraquinone moiety are presented, which were found identical in all the compounds, independent of alkyl chain length and platinum coordination. Apparently, there is little electronic interaction between platinum and the anthraquinone moiety. The predominantly red fluorescence emission ranges from 600 to 850 nm, with a maximum at 665 nm. The maxima in the excitation spectra at 586 nm and 630 nm are in agreement with the visible light absorption spectra [34].

Fluorescence emission spectrum (excited at 580 nm) and excitation spectrum (emission measured at 700 nm) of PAQ2 in water
Uptake of the compounds
The time-dependent average fluorescence of all compounds inside the cell are compared in Fig. 2. As the fluorescent part of all molecules is identical, these quantified data show the difference in uptake amongst the compounds. Apparently, the platinum complexes are taken up less efficiently than the free ligands. Further, the compounds with the shortest linkers are taken up most efficiently in both the ligand and platinum complex series. This observation nicely correlates with the cytotoxicity data: the most cytotoxic compounds are taken up most efficiently.

Quantification of the mean fluorescence intensity per pixel inside A2780 cells as determined digitally from the fluorescence microscopic images. Fluorescence intensity is given in alufi (arbitrary units of fluorescence intensity)
Platinum-free intercalators
Phase contrast and fluorescence images of the cells incubated with the metal-free compounds AQ2, AQ3 and AQ4 are presented in Fig. 3a, b and c, respectively. The most apparent difference among these platinum-free compounds is that AQ3 and AQ4 appear to be taken up less efficiently by the cells. Further, AQ4 displays a more distinct localization in the cell membrane, which may be related to the relatively high lipophilicity of AQ4 (see Fig. 3c). Apparently, the molecules may enter the cell by diffusion through the cell membrane. Also, small vesicles with high fluorescence intensity are observed already in the first minutes of incubation; this may indicate an alternative uptake mechanism through endocytosis, although it may as well be explained by aggregation of the compounds in the cytosol.

Representative phase contrast (top) and corresponding fluorescent images (bottom) of A2780 human ovarian carcinoma cells after 20 (left), 60 (middle) and 150 min (right) of incubation with the ligands AQ2 (a), AQ3 (b) and AQ4 (c)
Already after 15 min a significant amount of fluorescence is observed in the cytosol, but very little fluorescence is found in the nucleus. Interestingly, most fluorescence is located near the nucleus, in an area that, from spatial considerations, could very well be the Golgi apparatus. Other fluorescent intercalators, such as doxorubicin and daunorubicin, are known to accumulate in the Golgi apparatus [46, 47, 48, 49].
In order to verify whether the fluorescence is indeed located in the Golgi apparatus, a co-localization experiment was carried out. All three anthraquinones AQ2, AQ3 and AQ4 were assumed to give similar results for the co-localization experiment, as from the fluorescence microscopy results in Fig. 3 it appears that the compounds mainly differ in uptake efficiency as the localization patterns over time appear to be similar. The compound AQ2 is taken up the most efficiently and thus gives the most pronounced and clear fluorescence results, and for that reason a co-localization study was performed only on AQ2. After 20 min of incubation with AQ2, a Golgi-selective stain, NBD C6-ceramide, was added to the cells. However, no co-localization of AQ2 with the Golgi-specific marker was observed (images not shown).
In 2 h, fluorescence is increased in the nucleus, mostly concentrated in the nucleoli that are visible in the phase contrast microscopic images as black dots within the nucleus. The nucleoli are distinct structures inside the nucleus where ribosomes are produced. In the nucleoli, chromosomal DNA is translated into rRNA, and concentrated clusters of rRNA genes, rRNA and ribosomal proteins are present.
After 24 h, fluorescence is still visible in the cytosol around the nucleus and the nucleoli within the nucleus. Interestingly, the majority of the cells only contain a single fused nucleolus after 24 h, whereas multiple numbers of nucleoli are observed in the control cells. As the number of nucleoli varies throughout the cell-division cycle, this may indicate that the treated cells are arrested at a certain stage in the cell cycle.
Dinuclear platinum complexes
Microscopic images of the uptake and processing of the dinuclear platinum complexes PAQ2, PAQ3 and PAQ4 are shown in Fig. 4a, b and c, respectively. In contrast to the platinum-free compounds, the localization of fluorescence in the nucleoli was not observed for the platinum complexes.

Representative phase contrast (top) and corresponding fluorescent images (bottom) of A2780 human ovarian carcinoma cells after 20 (left), 120 minutes (middle) and 24 hours (right) of incubation with the dinuclear platinum complexes PAQ2 (a), PAQ3 (b) and PAQ4 (c)
The observation that little fluorescence accumulated within the nucleus may indicate that only a fraction of the drug molecules bind to nuclear DNA. The DNA was extracted from A2780 cells after incubation with the dinuclear platinum complexes and cisplatin, which was used as a reference. The drug-to-base-pair ratio (r b) was determined by UV and FAAS. The results are presented in Fig. 5. The platinum complexes with anthraquinones show even a higher degree of DNA binding than cisplatin. PAQ2 binds to the nuclear DNA more effectively than PAQ3 and PAQ4, which is in agreement with the cytotoxicity data. Thus, a considerable fraction of drug molecules interact with genomic DNA. However, little fluorescence was observed in the nuclei of the cells during the incubation. The fluorescence of some intercalators is known to be quenched when the aromatic moiety forms π-stacks with the DNA nucleobases. An additional control experiment was performed to investigate this possibility. From Fig. 6 it becomes clear that fluorescence quenches when a solution of AQ2 is titrated with a solution of pBR322 plasmid dsDNA. Although still some fluorescence is observed, the intensity drops to less than 5% of the original peak area. Upon the addition of 10 base-pair equivalents of DNA, a slight bathochromic shift is observed. Identical observations were made for the other compounds. As the nucleus of the cell contains concentrated dsDNA, the lack of fluorescence may be explained by such a quenching phenomenon.

DNA binding (r b=drug-to-base pair ratio) of cisplatin and dinuclear platinum complexes PAQ2, PAQ3 and PAQ4 in A2780 cells after 1 h of incubation at 200 μM

Quenching of the anthraquinone fluorescence emission upon titration of a solution of AQ2 with pBR322 plasmid DNA at various ratios (AQ2:DNA base pairs)
As the platinum complexes are taken up less efficiently than the platinum-free compounds, the fluorescence in images taken after 15 min of incubation can only be observed inside the cell when exposure times are increased. Most likely, the positive charges on the platinum ions make uptake by diffusion through the outer membrane more difficult compared to the free ligands. After 2 h, more fluorescence is observed within the cells in the cytosol surrounding the nucleus, similarly to the platinum-free compounds. A co-localization experiment with the Golgi-selective stain NBD C6-ceramide was performed for PAQ2, and as in the case of AQ2 no co-localization was found (images not shown). In contrast, in an earlier study by some of us [35], cisplatin derivatives fluorescently labeled with carboxyfluoresceine-diacetate (CFDA) and dinitrophenyl (DNP) were reported to localize in the Golgi apparatus, as proven by Golgi-selective staining. The cisplatin-type complexes are, however, structurally different from the complexes presented in the current study, and the observations were reported for human osteosarcoma (U2-OS) cells. The results of the earlier study [35] can therefore not be compared in a straightforward manner with the results of the current study.
After 24 h of incubation, an interesting difference in cellular processing of the ligands and the platinum complexes was observed. A considerable fraction (>50%) of the cells incubated with the platinum compounds contained fluorescent material encapsulated in unusually large vesicles in the cytosol (see Fig. 7). Apparently, the cellular processing for the free intercalators and the dinuclear platinum complexes differs, as these vesicles were not at all observed for the metal-free compounds. All the cells that contained these vesicles also featured a single large nucleolus, indicating cell cycle arrest.

Fluorescent material contained in large vesicles in A2780 cells after 24 h of incubation with PAQ2
In order to investigate the nature of large vesicles containing the platinum complexes PAQ2, PAQ3 and PAQ4 after 24 h of incubation, staining with LysoTracker Green DND-26 was performed. The LysoTracker probes are fluorescent acidotropic probes with high selectivity for acidic organelles [50, 51]. After 24 h of incubation with PAQ2 (PAQ3 and PAQ4 were assumed to behave in a similar way), LysoTracker Green DND-26 was added, and after 30 min of incubation with this dye the images shown in Fig. 8 were recorded. Figure 8B clearly shows that PAQ2, emitting red light, is sequestrated in large vesicles in cytosol. Staining of lysosomes is shown in green (Fig. 8C). The co-localization of PAQ2 with the marker, specific for acidic organelles, suggests that the vesicles encapsulating the dinuclear platinum complexes with anthraquinones are lysosome-like compartments. It must be noted that it is not certain that after 24 h the fluorescent molecules are still bound to platinum, as platinum may have been partly subtracted from the complexes of this type by intracellular thiols like glutathione [52].

Combination of PAQ2 accumulation after 24 h of incubation and staining with LysoTracker probe specific for acidic organelles in living A2780 human ovarian carcinoma cells. A Phase-contrast image of a living A2780 cell. B PAQ2 (red) accumulation in the same cell, 24 h of incubation with PAQ2. Accumulation in large vesicles. C Staining with LysoTracker Green DND-26 (green), which possesses high selectivity for acidic compartments. D Superimposed representation of the images of PAQ2 (red) and LysoTracker Green DND-26 (green)
A co-localization experiment has proven that the large vesicles observed during cellular processing of the complexes PAQ2, PAQ3 and PAQ4 are acidic compartments. The earlier evidences for platinum accumulation in lysosomes are known in the literature [53, 54, 55, 56]. It is often accompanied by an increase in the number of lysosomes [53, 56, 57, 58], and our case is no exception: after 24 h of incubation with the dinuclear platinum complexes, the cytosol of the cells is full of vesicles. Platinum sequestration in acidic compartments is likely to be employed by the cells to extrude toxic compounds. Platinum complexes localized in lysosomes may potentially alter lysosomal function, increasing activity of various lysosomal enzymes such as β-glucuronidase and alkaline phosphatase [53, 54]. Thus, the complexes might be degraded in the vesicles before being excreted from the cell. Sulfur-containing proteins, that are very reactive towards platinum [52, 59, 60], may also be involved in encapsulating of the platinum–anthraquinone complexes in vesicles.
As in this study only the fluorescing anthraquinone moieties within the cell are monitored and platinum itself is not directly observed, it must be noted that the presented results do not prove that platinum is still attached to an anthraquinone in the vesicles. Earlier studies [52, 61] have shown that platinum is easily removed from alkylenediamine ligands in dinuclear complexes by glutathione (GSH), and it is to be expected that, during cellular processing, platinum is eventually removed from at least part of the fluorescent anthraquinone. Apparently, however, the anthraquinones coordinated by platinum are clearly processed through different pathways compared to the uncomplexed anthraquinones.
Abbreviations
- AQ2:
-
N,N′-bis(2-aminoethyl)-1,4-diaminoanthracene-9,10-dione
- AQ3:
-
N,N′-bis(3-aminoprop-1-yl)-1,4-diaminoanthracene-9,10-dione
- AQ4:
-
N,N′-bis(4-aminobut-1-yl)-1,4-diaminoanthracene-9,10-dione
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide
- PAQ2:
-
[{trans-PtCl(NH3)2}2(μ-AQ2)](NO3)2
- PAQ3:
-
[{trans-PtCl(NH3)2}2(μ-AQ3)](NO3)2
- PAQ4:
-
[{trans-PtCl(NH3)2}2(μ-AQ4)](NO3)2
- PBS:
-
phosphate buffered saline
References
Hambley TW (1997) Coord Chem Rev 166:181–223
Wong E, Giandomenico CM (1999) Chem Rev 99:2451–2466
Farrell N, Qu Y, Bierbach U, Valsecchi M, Menta E (1999) Cisplatin, chemistry and biochemistry of a leading anticancer drug. Wiley-VCH, Weinheim, pp 479–496
Farrell N (1999) J Inorg Biochem 74:23–23
Reedijk J (2003) Proc Natl Acad Sci USA 100:3611–3616
Di Blasi P, Bernareggi A, Beggiolin G, Piazzoni L, Menta E, Formento ML (1998) Anticancer Res 18:3113–3117
Pratesi G, Perego P, Polizzi D, Righetti SC, Supino R, Caserini C, Manzotti C, Giuliani FC, Pezzoni G, Tognella S, Spinelli S, Farrell N, Zunino F (1999) Br J Cancer 80:1912–1919
Roberts JD, Peroutka J, Farrell N (1999) J Inorg Biochem 77:51–57
Roberts JD, Beggiolin G, Manzotti C, Piazzoni L, Farrell N (1999) J Inorg Biochem 77:47–50
Manzotti C, Pratesi G, Menta E, Di Domenico R, Cavalletti E, Fiebig HH, Kelland LR, Farrell N, Polizzi D, Supino R, Pezzoni G, Zunino F (2000) Clin Cancer Res 6:2626–2634
Komeda S, Lutz M, Spek AL, Chikuma M, Reedijk J (2000) Inorg Chem 39:4230–4236
Qu Y, Rauter H, Fontes APS, Bandarage R, Kelland LR, Farrell N (2000) J Med Chem 43:3189–3192
Perego P, Gatti L, Caserini C, Supino R, Colangelo D, Leone R, Spinelli S, Farrell N, Zunino F (1999) J Inorg Biochem 77:59–64
Perego P, Caserini C, Gatti L, Carenini N, Romanelli S, Supino R, Colangelo D, Viano I, Leone R, Spinelli S, Pezzoni G, Manzotti C, Farrell N, Zunino F (1999) Mol Pharmacol 55:1108–1108
Jansen BAJ, van der Zwan J, den Dulk H, Brouwer J, Reedijk J (2001) J Med Chem 44:245–249
Rauter H, Di Domenico R, Menta E, Oliva A, Qu Y, Farrell N (1997) Inorg Chem 36:3919–3927
Cox JW, Berners-Price S, Davies MS, Qu Y, Farrell N (2001) J Am Chem Soc 123:1316–1326
Zee-Cheng RK-Y, Podrebarac EG, Menon CS, Cheng CC (1979) J Med Chem 22:501–505
Krapcho AP, Getahun Z, Avery KL, Vargas KJ, Hacker MP, Spinelli S, Pezzoni G, Manzotti C (1991) J Med Chem 34:2373–2380
Gibson D, Binyamin I, Haj M, Ringel I, Ramu A, Katzhendler J (1997) Eur J Med Chem 32:823–831
Gibson D, Mansur N, Gean KF (1995) J Inorg Biochem 58:79–88
Gibson D, Gean KF, Benshoshan R, Ramu A, Ringel I, Katzhendler J (1991) J Med Chem 34:414–420
Kitov S, Benshoshan R, Ringel I, Gibson D, Katzhendler J (1988) Eur J Med Chem 23:381–383
Gean KF, Benshoshan R, Ramu A, Ringel I, Katzhendler J, Gibson D (1991) Eur J Med Chem 26:593–598
Mikata Y, Yokoyama M, Mogami K, Kato M, Okura I, Chikira M, Yano S (1998) Inorg Chim Acta 279:51–57
Mikata Y, Mogami K, Kato M, Okura I, Yano SB (1997) Bioorg Med Chem Lett 7:1083–1086
Ceci E, Cini R, Karaulov A, Hursthouse MB, Maresca L, Natile G (1993) J Chem Soc Dalton Trans 2491–2497
Cullinane C, Wickham G, McFadyen WD, Denny WA, Palmer BD, Phillips DR (1993) Nucleic Acids Res 21:393–400
Goodgame DML, Page CJ, Stratford IJ (1991) Transition Met Chem 16:223–229
Bowler BE, Ahmed KJ, Sundquist WI, Hollis LS, Whang EE, Lippard SJ (1989) J Am Chem Soc 111:1299–1306
Murray V, Motyka H, England PR, Wickham G, Lee HH, Denny WA, McFadyen WD (1992) J Biol Chem 267:18805–18809
Temple MD, McFadyen WD, Holmes RJ, Denny WA, Murray V (2000) Biochemistry 39:5593–5599
Jamieson ER, Lippard SJ (1999) Chem Rev 99:2467–2498
Smith PJ, Desnoyers R, Blunt N, Giles Y, Patterson LH, Watson JV (1997) Cytometry 27:43–53
Molenaar C, Teuben JM, Heetebrij RJ, Tanke HJ, Reedijk J (2000) J Biol Inorg Chem 5:655–665
Qu Y, Farrell N (1992) Inorg Chem 31:930–932
Jansen BAJ, van der Zwan J, Reedijk J, den Dulk H, Brouwer J (1999) Eur J Inorg Chem 1429–1433
Kaufman EB, Cowan DO (1968) Inorg Synth 7:242–245
Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, Abbott BJ, Mayo JG, Shoemaker RH, Boyd MR (1988) Cancer Res 48:589–601
Krapcho AP, Getahun Z, Avery KJ (1990) Synth Commun 20:2139–2146
Zhen WP, Link CJ, O’Connor PM, Reed E, Parker R, Howell SB, Bohr VA (1992) Mol Cell Biol 12:3689–3698
Kelland LR, Barnard CFJ, Mellish KJ, Jones M, Goddard PM, Valenti M, Bryant A, Murrer BA, Harrap KR (1994) Cancer Res 54:5618–5622
Holford J, Sharp SY, Murrer BA, Abrams M, Kelland LR (1998) Br J Cancer 77:366–373
Kelland LR, Barnard CFJ, Evans IG, Murrer BA, Theobald BRC, Wyer SB, Goddard PM, Jones M, Valenti M, Bryant A, Rogers PM, Harrap KR (1995) J Med Chem 38:3016–3024
Kalayda GV, Jansen BAJ, Wielaard P, Molenaar C, Tanke HJ, Reedijk J (2004) J Biol Inorg Chem 9:(accompanying paper)
Huxham IM, Barlow AL, Lewis AD, Plumb J, Mairs RJ, Gaze MN, Workman P (1994) Int J Cancer 59:94–102
Arancia G, Calcabrini A, Meschini S, Molinari A (1998) Cytotechnology 27:95–111
Bour-Dill C, Gramain MP, Merlin JL, Marchal S, Guillemin F (2000) Cytometry 39:16–25
Beyer U, Rothen-Rutishauser B, Unger C, Wunderli-Allenspach H, Kratz F (2001) Pharm Res 18:29–38
Racoosin EL, Swanson JA (1993) J Cell Biol 121:1011–1020
Kim JH, Lingwood CA, Williams DB, Furuya W, Manolson MF, Grinstein S (1996) J Cell Biol 134:1387–1399
Jansen BAJ, Brouwer J, Reedijk J (2002) J Inorg Biochem 89:197–202
Aggarwal SK (1993) J Histochem Cytochem 41:1053–1073
Litterst CL (1984) Agents Actions 15:520–524
Dietel M (1991) Pathol Res Pract 187:892–905
Edwards PG, Kendall MD, Morris IW (1991) Scanning Microsc 5:797–810
Cece R, Petruccioli MG, Cavaletti G, Barajon I, Tredici G (1995) Histol Histopathol 10:837–845
Muenchen HJ, Aggarwal SK (1998) Anticancer Res 18:2631–2636
Berners-Price SJ, Kuchel PW (1990) J Inorg Biochem 38:305–326
Reedijk J (1999) Chem Rev 99:2499–2510
Oehlsen ME, Qu Y, Farrell N (2003) Inorg Chem 42:5498–5506
Acknowledgements
The authors are indebted to the EU for a grant as a Training Host Institute in the EU programme Human Capital and Mobility. Also support and sponsorship by COST Actions D20/0001/00, D20/0002/00 and D20/003/01 (biocoordination chemistry) is kindly acknowledged. The authors wish to thank Johnson Matthey (Reading, UK) for their generous gift of K2PtCl4. This work has been performed under auspices of the joint BIOMAC Research Graduate School of Leiden University and Delft University of Technology.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jansen, B.A.J., Wielaard, P., Kalayda, G.V. et al. Dinuclear platinum complexes with N,N′-bis(aminoalkyl)-1,4-diaminoanthraquinones as linking ligands. Part I. Synthesis, cytotoxicity, and cellular studies in A2780 human ovarian carcinoma cells. J Biol Inorg Chem 9, 403–413 (2004). https://doi.org/10.1007/s00775-004-0539-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-004-0539-y