
Abstract
Antimicrobial peptides (AMPs) play a key role in the defence mechanism of living organisms against microbial pathogens, displaying both bactericidal and immunomodulatory properties. They are considered as a promising alternative to the conventional antibiotics towards which bacteria are becoming highly resistant. Recently, a derivative of the frog skin AMP esculentin-1a, esculentin-1a(1–21)NH2 [Esc(1–21)], showed a strong and fast membranolytic activity against Gram-negative bacteria but with a lower efficacy against Gram-positive ones. Here, with the aim to increase the α-helicity of Esc(1–21) and the expected potency against Gram-positive bacteria, we designed an analog bearing three α-aminoisobutyric acid (Aib) residues at positions 1, 10, and 18 of its primary structure. We demonstrated that the incorporation of Aib residues: (1) promoted the α-helix conformation of Esc(1–21), as confirmed by circular dichroism and two-dimensional nuclear magnetic resonance spectroscopies; (2) was sufficient to make this analog more active than the parent peptide against several Gram-positive bacterial strains without affecting its activity against Gram-negative bacteria; and (3) resulted to be devoid of toxic effect toward epithelial cells at the active antimicrobial concentrations. These results suggest that replacement of L-amino acids with Aib residues has beneficial effects on the structure and properties of the membrane-active peptide Esc(1–21), making it a better candidate for the design and development of selective drugs against Gram-positive bacteria.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ribosomally made antimicrobial peptides (AMPs) are produced by all species of life throughout the evolutionary scale as principal components of their innate defence system against invading microorganisms (Gonzalez-Navajas et al. 2014; Mangoni and Shai 2011; Mookherjee and Hancock 2007; Nicolas and Mor 1995). Furthermore, they are endowed with immunomodulatory properties (Choi et al. 2012; Hemshekhar et al. 2016) which have led to the more appropriate designation of “host-defence peptides” (Hancock et al. 2016; Mansour et al. 2014). More specifically, amphibian skin dermal glands, controlled by sympathetic nerves, are among the richest sources of biologically active peptides with pharmacological and antimicrobial activities (Chen et al. 2003; Conlon 2011; Erspamer 1971; Haslam et al. 2014; Konig et al. 2014; Mangoni et al. 2015). They are stored within granules and released on the skin surface by a holocrine mechanism, upon alarm or physical injury (Mangoni et al. 2001, 2007). Each frog species produces its own unique set of AMPs encompassing families of 2–100 closely related members (Mangoni 2006). Esculentins-1 are a class of frog skin AMPs, characterized by a 46 amino acids primary structure and a broad range of antimicrobial activity (Gamberi et al. 2007; Mangoni et al. 2003; Ponti et al. 2003; Simmaco et al. 1994). Studies on their mode of action pointed out the bacterial membrane as the major target. Esculentins-1 possess features common to most linear AMPs, i.e., an overall positive charge at neutral pH and a considerable proportion of hydrophobic residues (Simmaco et al. 1994). These properties are instrumental in allowing an electrostatic interaction between the cationic AMP and the negatively charged components of the microbial cell surface followed by the peptide’s folding into an amphiphilic structure, with a resulting perturbation of the cell membrane permeability and hence cell death (Ganz and Lehrer 1998; Haney et al. 2010; Lohner and Blondelle 2005; Shai 2002). Importantly, in contrast with the conventional antibiotics that interfere with biological events by processes involving specific recognition of chiral targets (Bai et al. 2011; Levy 2002; Savjani et al. 2009), the mechanism of action underlying the killing activity of AMPs is generally based on the physical disruption of the target cell membrane, thus limiting the induction of microbial resistance (Hancock and Rozek 2002; Lohner 2016). Indeed, to become resistant to AMPs, microbes should drastically change the composition of their membrane, an event that could not be achieved without causing a significant harm to the microorganism itself (Mangoni 2006; Mangoni and Shai 2011). It is worth recalling that the membrane of mammalian cells is much richer in zwitterionic phospholipids as compared with that of microbial cells and this difference is one of the major reasons accounting for the preferential activity of AMPs towards bacterial and fungal cells (Epand and Vogel 1999).
Previous studies reported that the N-terminal derivative of esculentin-1a, esculentin-1a(1–21)NH2, [Esc(1–21)] corresponding to its first 20 amino acids followed by an amidated Gly residue (H-Gly-Ile-Phe-Ser-Lys-Leu-Ala-Gly-Lys-Lys-Ile-Lys-Asn-Leu-Leu-Ile-Ser-Gly-Leu-Lys-Gly-NH2) adopts an alpha-helical conformation in a membrane-mimicking environment and retains the antimicrobial activity of the full-length peptide esculentin-1a (Islas-Rodriguez et al. 2009; Gamberi et al. 2007; Ghosh et al. 2016). More recently, Esc(1–21) was shown to display a fast membranolytic activity against both planktonic and biofilm forms of the multi-drug resistant (MDR) opportunistic Gram-negative bacterium Pseudomonas aeruginosa (Breidenstein et al. 2011; Drenkard and Ausubel 2002; Kolar et al. 2015; Luca et al. 2013; Uccelletti et al. 2010). This bacterium has the ability to colonize both inert surfaces (such as those of medical devices, e.g., contact lenses) and biological tissues, forming biofilm communities (Hoiby et al. 2011; Parsek and Tolker-Nielsen 2008; Rybtke et al. 2015) which can easily lead to acute and chronic infections, including otitis, pneumonia, and keratitis (Abbouda et al. 2016; Bodey et al. 1983). However, a lower efficacy has been shown by Esc(1–21) against Gram-positive bacteria (Kolar et al. 2015).
In this connection, with the aim of enlarging the spectrum of activity of Esc(1–21) especially against Gram-positive bacteria as well as its biostability to proteases, we explored the effects of the incorporation of α-aminoisobutyric acid (Aib) residues into the peptide sequence. When inserted into the primary structure of peptides, this strongly helicogenic, non-coded, Cα-tetrasubstituted α-amino acid is expected to increase the α-helical content of the molecule (Karle and Balaram 1990; Toniolo et al. 2001) and potentially confers it a higher resistance against enzymatic degradation (De Zotti et al. 2009, 2012; Yamaguchi et al. 2003). Furthermore, it was previously demonstrated that a stabilized α-helical structure is an essential requirement to enhance the microbicidal activity of a peptide against Gram-positive bacteria and fungi (Giangaspero et al. 2001). In this work, we report on the synthesis of an analog of Esc(1–21), bearing three Aib residues at sequence position 1, 10, and 18 {[Aib1,10,18]-Esc(1–21)}; its structural characterization in different environments, by circular dichroism (CD) and two-dimensional nuclear magnetic resonance spectroscopies (2D-NMR) techniques, as well as its biological activity, and compared these results with those of the parent peptide Esc(1–21).
Materials and methods
Materials
Fmoc-amino acids were supplied from Novabiochem (Merck Biosciences, La Jolla, CA, USA), and all other amino-acid derivatives and reagents for peptide synthesis were purchased from Sigma-Aldrich (St. Louis, MO, USA). 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) was purchased from GLS (Shanghai, China). Trypsin–EDTA was purchased from Invitrogen (Life-Technologies Europe, Monza, Italy); 3(4,5-dimethylthiazol-2yl)2,5-diphenyltetrazolium bromide (MTT) was from Sigma-Aldrich (St. Luis, MO). Dulbecco’s modified Eagle’s medium (DMEM), heat-inactivated fetal bovine serum (FBS), glutamine, gentamycin, and penicillin/streptomycin were from Euroclone (Milan, Italy). Esc(1–21) was purchased from Selleck Chemicals (Houston, TX, USA) and purified according to (Di Grazia et al. 2015a, b).
Synthesis of [Aib1,10,18]-Esc(1–21)
Assembly of the peptide on the Advanced ChemTech (Louisville, KY, USA) 348 Ω peptide synthesizer was performed on a 0.06-mmol scale by the FastMoc methodology [HBTU, HOBt, DIEA, single acylation protocol, 45 min coupling time, N,N-dimethylformamide (DMF) as the solvent], starting with Rink Amide MBHA resin (Iris Biotech, Marktredwitz, Germany) (95 mg, loading 0.65 mmol g−1). The deprotection of the Fmoc group was performed with a 20 % piperidine solution in DMF in two steps of 5 and 15 min, respectively. The coupling steps involving Aib residues, carried out in presence of HATU, were repeated twice. Boc and tBu side-chain protections were used for Lys and Ser residues, respectively. Cleavage of the peptide from the resin, concomitantly with side-chain deprotections, was achieved by treatment with trifluoroacetic acid (TFA)/triisopropylsilane (TIS)/water (95:2.5:2.5 v/v). The crude peptide was purified by reverse-phase flash chromatography using a Biotage Isolera Prime (Uppsala, Sweden) purification system. The chromatrographically homogeneous, final peptide was characterized by electrospray ionization mass spectrometry (ESI–MS) and NMR.
Microorganisms
The microorganisms used for the antimicrobial assays were the reference Gram-negative bacteria Acinetobacter baumannii ATCC 19606, Escherichia coli D21, E. coli ATCC 25922, Pseudomonas aeruginosa ATCC 27853, Yersinia pseudotuberculosis YPIII, and the Gram-positive bacteria Bacillus megaterium Bm11, Staphylococcus epidermidis ATCC 12228, as well as the clinical isolates Staphylococcus aureus 6938, Staphylococcus capitis 1, Staphylococcus epidermidis 21, and Staphylococcus hominis 1. In addition, two Candida strains were employed: the reference Candida albicans ATCC 10231 and C. guillier mondii from the frog natural flora (Mangoni et al. 2001).
Antimicrobial assay
Susceptibility testing was performed by adapting the microbroth dilution method outlined by the Clinical and Laboratory Standards Institute, using sterile 96-well plates (Falcon NJ, USA). The bacterial growth was aseptically measured by absorbance at 590 nm with a spectrophotometer (UV-1700 Pharma Spec Shimadzu, Tokyo, Japan). Aliquots (50 μl) of bacteria in mid-log phase at a concentration of 2 × 106 colony-forming units (CFU)/ml in culture medium (Mueller–Hinton, MH) were added to 50 μl of MH broth containing the peptide in serial twofold dilutions ranging from 64 to 0.25 μM. Inhibition of microbial growth was visually observed, after 18 h incubation at 37 °C. Antibacterial activity was expressed as the minimal inhibitory concentration (MIC), the concentration of peptide causing 100 % inhibition of microbial growth. The same procedure was followed with yeasts in Winge medium (Valenti et al. 1985) using a final cell concentration of 3.5 × 104 CFU/ml and an incubation time of 18 h at 30 °C.
Cell cultures
The human type II alveolar epithelial cells (A549 cell line from the American Type Culture Collection) and the human immortalized keratinocytes (HaCaT) cell line were employed. Cells were cultured in DMEM containing 10 % heat-inactivated fetal bovine serum (FBS) and supplemented with l-glutamine (2 mM or 4 mM for A549 or HaCaT cells, respectively) and antibiotics (0.1 mg/ml of penicillin and streptomycin for A549 cells; 0.05 mg/ml of gentamicin for HaCaT cells) at 37 °C and 5 % CO2 in 25-cm2 flasks.
Peptides’ effect on cell viability
The effect of both peptides on the viability of mammalian cells was determined by the inhibition of MTT reduction to insoluble formazan, by mitochondrial reductases. Cells suspended in the corresponding culture medium supplemented with glutamine and 2 % FBS without antibiotics were plated in triplicate wells of a microtiter plate, at 4 × 104 cells/well. After overnight incubation at 37 °C in a 5 % CO2 atmosphere, the medium was replaced with 100 μl fresh serum-free medium containing the peptides at different concentrations. The plate was incubated for 2 h or 24 h at 37 °C in a 5 % CO2 atmosphere (Paiva et al. 2012). Then, the culture medium was removed and replaced with Hank’s buffer (136 mM NaCl; 4.2 mM Na2HPO4; 4.4 mM KH2PO4; 5.4 mM KCl; 4.1 mM NaHCO3, pH 7.2, supplemented with 20 mM d-glucose) containing 0.5 mg/ml MTT. After 4 h incubation, the formazan crystals were dissolved by adding 100 μl of acidified isopropanol and viability was determined by absorbance measurements at 570 nm using a microplate reader (Infinite M200; Tecan, Salzburg, Austria). Cell viability was calculated with respect to the control (cells not treated with peptide). The percentage of viable cells was calculated according to the formula:
where the blank is given by samples without cells and not treated with the peptide.
Circular dichroism spectroscopy
The CD spectra were measured on a Jasco (Hachioji City, Japan) model J-715 spectropolarimeter equipped with a Haake thermostat (Thermo Fisher Scientific, Waltham, MA, USA). Milli-Q grade water, spectrograde methanol, and TFE (Acros Organic, Geel, Belgium) were used as solvents. Peptide concentrations were determined by UV absorption at 254 nm. For each spectrum, a total of eight scans were averaged. Baselines were corrected by subtracting the solvent contribution. Fused quartz cell of 0.1-mm path length (Hellma, Mühlheim, Germany) was used. For the experiments carried out in mixed solvents, the proper ratio of the individual solutions at the same concentration was mixed. The values are expressed in terms of [θ]T, the total molar ellipticity (deg × cm2 × dmol− 1).
Nuclear magnetic resonance spectrometry
Samples for NMR spectrometry were dissolved in TFE-d2 solution (peptide concentrations: about 1 and 1.5 mM, respectively, for Esc(1–21) and its Aib1,10,18 analog. The spectra were recorded at 298 K. All NMR experiments were performed on a Bruker Avance DMX-600 spectrometer using the TOPSPIN 1.3 software package. Presaturation of the H2O signal was obtained using a WATERGATE gradient program. All homonuclear spectra were acquired by collecting 512 experiments, each one consisting of 64–80 scans and 2 K data points. The spin systems of the protein amino-acid residues were identified using the standard DQF-COSY (Rance et al. 1983) and CLEAN-TOCSY (Bax and Davis 1985; Griesinger et al. 1988) spectra. In the latter case, the spin-lock pulse sequence was 70 ms long. NOESY experiments were used for sequence-specific assignment (Wüthrich 1986), the mixing time used was 150 ms to avoid spin-diffusion problems.
Statistical analysis
Data were collected from three independent experiments. Quantitative data are expressed as the mean ± SEM. Statistical analysis was performed using the two-way analysis of variance (ANOVA), with the PRISM software (GraphPad, San Diego, CA, USA). Differences were considered to be statistically significant for p < 0.05. The levels of statistical significance are indicated in the legend to figures.
Results and discussion
Synthesis
We synthesized an analog of Esc(1–21) in which we inserted three Aib residues at positions 1, 10, and 18. The choice of the sequence positions of Esc(1–21) to be replaced by Aib was based on the following considerations: (1) the placement of a non-coded Aib residue at position 1 might prevent proteolytic degradation by aminopeptidases, whereas an amidated peptide C-terminus is known to confer protection against carboxypeptidases (Rink et al. 2010; Veber and Freidinger 1985), thus suggesting that the presence of a C-terminal Aib would not be necessary. Concerning the protection by endopeptidases, the introduction of a few additional Aib replacements, possibly quite evenly distributed along the primary structure, is expected to be (at least to some extent) beneficial. (2) The helix-promoting capabilities of Aib are more effective when this residue is placed internal to the peptide sequence, where it can display its influence on both the preceding and the following residues in the primary sequence. (3) A helical wheel plot of the primary structure of Esc(1–21), where residues are arranged according to an ideal α-helical folding, is shown in Fig. 1a. In this putative fully α-helical conformation, two faces can be identified, one possessing a more pronounced hydrophobic character and the other a more hydrophilic one. However, strongly hydrophobic residues, such as Ile2, Leu6, and Ile16, are located on the same face occupied by the five charged Lys residues. (4) It is worth recalling that in naturally occurring Aib-rich amphipathic helical peptides of fungal origin known as peptaibiotics (Toniolo and Brückner 2009), the Aib residues are, in general, located within the hydrophobic face but also at its boundary with the hydrophilic one. The most striking example of this latter disposition is provided by the lipopeptide trichogin (Toniolo et al. 1994). All together, these observations suggested us to place three Aib residues in the Esc(1–21) sequence; two of them at positions 1 and 18 (both as a replacement for Gly), and one in substitution for Lys10, i.e., at the boundary between the hydrophilic and hydrophobic faces (Fig. 1b). As a result of this latter replacement, the overall net charge decreases by one unit if compared with that of the parent peptide, and the overall hydrophilic/hydrophobic profile becomes slightly modified. We principally preferred to give priority to the increase of the helical propensity and proteolytic stability of the central part of the sequence expected as a result of the Lys10 → Aib10 substitution, even if accompanied by the potentially unfavorable effects outlined above. For the solid-phase peptide synthesis of [Aib1,10,18]-Esc(1–21), we exploited a well-established protocol concerning the use of a strong activating agent, HATU, in the coupling reactions involving Aib residues (De Zotti et al. 2012). The synthesis was performed on a Rink amide MBHA resin using an Fmoc Nα protection protocol as described in “Materials and methods”. The cleavage of the peptide from the resin was achieved using a mixture of TFA/TIS/H2O. The crude peptide was obtained in 80 % yield with a purity of 85 %, as evidenced by RP-HPLC. Reverse-phase flash chromatography allowed the isolation of the peptide with 97 % purity.

Helical wheel plots of the primary structure of Esc(1–21) (a) and its Aib1,10,18 analog (b)
H-Aib1-Ile-Phe-Ser-Lys-Leu-Ala-Gly-Lys-Aib10-Ile-Lys-Asn-Leu-Leu-Ile-Ser-Aib18-Leu-Lys-Gly-NH2
MW (calcd for C104H185N27O24) | MW (experimental) | t r (min) | Purity | |
|---|---|---|---|---|
[Aib1,10,18]-Esc(1–21) | 2196.41 | 2196.40 | 9.38a | 97 % |
Antimicrobial activity
The activity of [Aib1,10,18]-Esc(1–21) against different microorganisms, including Gram-negative, Gram-positive bacteria, and yeasts, was tested by the microdilution broth assay to determine the MIC. In comparison with the parent peptide, the incorporation of Aib residues sharply increases the activity of the peptide against Gram-positive bacteria, as shown by its lower MICs in Table 1. More specifically, the MIC of the analog carrying Aib residues is eightfold lower against S. epidermidis strains or 16-fold lower against S. capitis 1. Furthermore, the antibacterial activity of [Aib1,10,18]-Esc(1–21) becomes even stronger against S. aureus, with a 32-fold lower MIC than that of Esc(1–21). Interestingly, in line with what previously found for the de novo designed P19 peptide (Giangaspero et al. 2001), the presence of Aib residues within the peptide sequence does not significantly affect the activity of the peptide against Gram-negative bacteria, as indicated by the same MIC values to those of the parent peptide (Table 1), with the exception of E. coli ATCC 25922 towards which the analog results to be only twice as powerful as Esc(1–21). In addition, the anti-yeast activity is not significantly influenced by the presence of Aib residues, and the MIC against Candida strains is equal or twofold lower than that of Esc(1–21). It is very well known that the cell selectivity of AMPs is governed by several biophysical and biochemical factors, including not only the peptide’s cationicity, amphipathicity, hydrophobicity, chain length, helicity, and oligomeric state, but also the properties of the target cell surface (Glukhov et al. 2005; Matsuzaki 2009). In the last 15 years, several studies reported that an amphipathic structure is a primary requirement for AMPs to be able to kill Gram-positive bacteria and fungi, while Gram-negative bacteria remain susceptible to both non-helical and scrambled peptides (Dathe et al. 1996, 1997; Giangaspero et al. 2001). However, it is not easy to provide an unequivocal explanation for the difference in the activity profile of Esc(1–21) and its Aib-containing analog against the three classes of microorganisms. The different lipid composition existing between the membrane of Gram-positive and Gram-negative bacteria or fungi certainly plays a crucial role in determining the feasibility of the peptide’s insertion into the hydrophobic core of the phospholipid bilayer, which results in membrane destabilization/perturbation and microbial death (Epand et al. 2007; Epand and Vogel 1999). In addition, differences in the cell wall architecture of the target microorganism would contribute to variations in the peptides’ antimicrobial efficacy. Indeed, before reaching the target cytoplasmic membrane, AMPs need to interact with lipopolysaccharides of the outer membrane that surrounds the cell wall in Gram-negative bacteria (Bhunia et al. 2009, 2010; Domadia et al. 2010). Differently, a thicker peptidoglycan or a glucan–rich layer is present in the cell wall of Gram-positive bacteria or fungi, respectively (Free 2013; Schaffer and Messner 2005). Presumably, according to the previous studies (Giangaspero et al. 2001), more stringent structural requirements of a peptide may favor its translocation through the peptidoglycan barrier of Gram-positive bacteria into the cytoplasmic membrane. This observation may at least in part justify the lower MIC values found for the more helical [Aib1,10,18]-Esc(1–21) compared with the parent Esc(1–21) against these bacterial strains.
Peptides’ effect on viability of mammalian cell lines
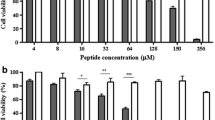
The effect of Aib introduction within the primary structure of Esc(1–21) on the viability of eukaryotic cells was studied by the MTT assay on different types of mammalian epithelial cell lines: the human alveolar lung epithelial A549 cells and the human keratinocyte HaCaT cells. As reported in Fig. 2a, viability of A549 cells after 2 h exposure to [Aib1,10,18]-Esc(1–21) at concentrations 2–4 µM is not significantly reduced and there is no significant difference between the two peptides.

Peptides’ effect on the viability of A549 cells (a, c) or HaCaT cells (b, d). Cells were plated in wells of a microtiter plate, at 4 × 104 cells/well in culture medium, as described in the Experimental section. After overnight incubation at 37 °C in a 5 % CO2 atmosphere, the medium was replaced with 100 μl fresh medium supplemented with the peptides at different concentrations. After 2 h (a, b) or 24 h (c, d) of peptide treatment, cell viability was determined by the MTT reduction to insoluble formazan. Cell viability is expressed as percentage with respect to the control (cells not treated with the peptide). Data points represent the mean of triplicate samples ± SEM. The data of the wild-type peptide Esc(1–21) on HaCaT cells were taken from our previous work (Di Grazia et al. 2015a). The levels of statistical significance between the two peptides are: ***p < 0.001; ****p < 0.0001
Similarly, when [Aib1,10,18]-Esc(1–21) is analyzed against HaCaT cells (Fig. 2b), any marked reduction in the number of metabolically active cells is obtained within the peptide concentrations of 2–4 μM compared with the harmless parent peptide. These findings suggest that [Aib1,10,18]-Esc(1–21) is not toxic to mammalian cells when used at its growth inhibitory concentrations against Gram-positive bacteria (Table 1). Note, however, that when the Aib-containing analog is tested at higher concentrations, i.e., 16, 32, and 64 µM against A549 cells, cell viability decreases to ~60, 20, and 5 %, respectively (Fig. 2a) or even further when the peptide is assayed against keratinocytes (Fig. 2b). As reported in Fig. 2c and d, the cytotoxicity of [Aib1,10,18]-Esc(1–21) is only slightly increased after a long-term peptide treatment (24 h), indicating that an irreversible damage has been caused to the cells.
Note that the higher cytotoxicity of the more helical Aib-containing Esc(1–21) is in agreement with the previous findings showing that the ease of α-helix formation and stability are important factors for the mammalian membrane perturbation and cell lysis (Gazit et al. 1994; Pouny et al. 1992; Shai and Oren 1996; Strahilevitz et al. 1994).
Circular dichroism
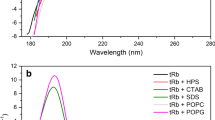
Far-UV CD spectra of Esc(1–21) and its Aib1,10,18 analog were acquired in three different solvents: water, TFE, and 100 mM sodium dodecyl sulfate (SDS) aqueous solution (Fig. 3a, b). In water, both peptides exhibit a random coil structure, while in TFE and micellar SDS aqueous solution, they adopt an overall helical conformation. In both membrane-mimicking environments, each spectrum shows two negative maxima near 205 and 222 nm and one positive maximum at 195 nm, indicative of a right-handed helical conformation for both peptides (Beychock 1967). The ellipticity ratio R = [θ]222/[θ]205 calculated in TFE and SDS solution evidenced a predominant α-helical conformation for both analogs (Manning and Woody 1991).

Far-UV CD spectra of: a Esc(1–21) and b [Aib1,10,18]-Esc(1–21) in three different environments: water, TFE, and 100 mM SDS solution; c Esc(1–21) and d [Aib1,10,18]-Esc(1–21) in water, 20 % TFE, 50 % TFE, and 100 % TFE (peptide concentration 1 mM)
R SDS | R TFE | |
|---|---|---|
Esc(1–21) | 0.76 | 0.76 |
[Aib1,10,18]-Esc(1–21) | 0.92 | 0.75 |
The CD results outlined above would seem to suggest that the two compounds are conformationally similar. However, a significant difference emerges from the analysis of the CD spectra collected in water/TFE mixtures of varying composition (Fig. 3c, d). Indeed, for each peptide, the set of spectra is characterized by an isodichroic point at 203 nm, consistent with a two-state transition from the unordered conformation in water to the helical structure in 100 % TFE. The helical content of Esc(1–21) increases sharply with increasing TFE percentage from 20 to 50 % and to a lower extent from 50 to 100 % TFE (Fig. 3c), whereas the Aib-containing analog appears to be much more helical than the parent peptide already at 20 % TFE, and its CD spectra vary little at higher TFE percentages (Fig. 3d).
Nuclear magnetic resonance analysis
The 2D-NMR spectra of Esc(1–21) and its Aib1,10,18 analog were recorded in TFE solution. The proton resonances were fully assigned following the Wüthrich procedure (Wüthrich 1986).
The NOESY spectra in TFE solution of both peptides evidenced the presence of most of NHi–NHi+1 sequential cross peaks, indicative of the occurrence of helical structure (Fig. 4), thus confirming the information obtained from CD spectra.

Fingerprint region of the H/H-NOESY spectrum of Esc(1–21) (a) and [Aib1,10,18]-Esc(1–21) (b) (600 MHz, 1.1 and 1.4 mM, respectively, in TFE-d2 solution, 298 K). The CαHi → NHi+2 (red), CαHi → NHi+3 (green), and CαHi → NHi+4 (blue) cross peaks, diagnostic of helical conformation, are highlighted
The NOESY fingerprint region of Esc(1–21) shows the CαHi → NHi+2 and CαHi → NHi+3 cross peaks, diagnostic of helical conformation (Fig. 4a), even if a CαHi → NHi+4 cross peak, characteristic of α-helical structure could be detected only in the Phe3–Ala7 segment (although the extensive overlapping of the signals might hamper their detection in other portions of the sequence).
The fingerprint region of the NOESY spectrum of the Aib1,10,18 analog results better resolved (Fig. 4b). A number of connectivities are necessarily missing, because the quaternary Aib residues (at sequence positions 1, 10, and 18) lack the α hydrogen atom. Nevertheless, in addition to the CαHi → NHi+2 and CαHi → NHi+3 cross peaks (the latter in a larger number if compared to the parent peptide), two CαHi → NHi+4 are also evident at the level of the Phe3–Ala7 and Leu15–Leu19 segments.
A comparison of the conformationally relevant signatures extracted from the NOESY spectra of Esc(1–21) and its Aib1,10,18 analog is reported in Fig. 5. Overall, in the C-terminal portion of the Aib1,10,18 analog, the number of connectivities consistent with a helical conformation is more abundant than in the corresponding region of Esc(1–21). Therefore, both peptides are largely helical, but the introduction of Aib residues appears to increase the population and stability of the helix in the otherwise more flexible C-terminal domain of Esc(1–21).

Summary of the significant interresidue NOESY cross peaks for Esc(1–21) (a) and [Aib1,10,18]-Esc(1–21) (b) in TFE-d2 solution. Peptide concentration 1.1 and 1.4 mM, respectively
Conclusions and perspectives
In this work, we demonstrated that the increased alpha-helical content of Esc(1–21), obtained by incorporation of three non-proteinogenic Aib residues at positions 1, 10, and 18 (as shown by CD and NMR studies) is sufficient to provoke a dramatic increase in the peptide’s activity against Gram-positive bacteria without significantly increasing its toxicity towards epithelial cells at antimicrobial concentrations. This suggests that the Aib designed analog is a better candidate than the wild-type peptide for the development of a new drug against Gram-positive bacterial infections, such as those associated with the human skin or the lung (Lee et al. 2015; Soufi and Soufi 2016). Nevertheless, considering the toxicity of this analog at concentrations higher than the MICs, it would also be useful to develop this peptide for further applications, such as those related to the usage of friendly biocides against Gram-positive bacterial communities on metal surfaces in marine engineering systems, e.g., pipelines of the offshore oil and gas industry, to prevent substantial corrosion problems and contamination of agricultural lands (Godwin and Akpan 2014; Schwermer et al. 2008).
Abbreviations
- CD:
-
Circular dichroism
- DIEA:
-
N,N-Diisopropylethylamine
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- FBS:
-
Heat-inactivated fetal bovine serum
- HATU:
-
1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- HBTU:
-
N,N,N′,N′-Tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate
- HOBt:
-
1-Hydroxybenzotriazole
- MBHA:
-
4-Methylbenzhydrylamine
- MH:
-
Mueller–Hinton
- MTT:
-
3(4,5-Dimethylthiazol-2yl)2,5-diphenyltetrazolium bromide
- NMR:
-
Nuclear magnetic resonance
- SDS:
-
Sodium dodecylsulfate
- TIS:
-
Triisopropylsilane
- TFA:
-
Trifluoroacetic acid
- TFE:
-
Trifluoroethanol
References
Abbouda A, Abicca I, Alio JL (2016) Infectious keratitis following corneal crosslinking: a systematic review of reported cases: management, visual outcome, and treatment proposed. Semin Ophthalmol 31:485–491
Bai H, Zhou Y, Hou Z, Xue X, Meng J, Luo X (2011) Targeting bacterial RNA polymerase: promises for future antisense antibiotics development. Infect Disord Drug Targets 11:175–187
Bax A, Davis DG (1985) MLEV-17-based two dimensional homonuclear magnetization transfer spectroscopy. J Magn Reson 65:355–360
Beychock S (1967) Circular dichroism of poly-α-amino acids and proteins. In: Fasman GD (ed) Poly-α-amino acids: protein models for conformational studies. Dekker, New York, pp 293–337
Bhunia A, Ramamoorthy A, Bhattacharjya S (2009) Helical hairpin structure of a potent antimicrobial peptide MSI-594 in lipopolysaccharide micelles by NMR spectroscopy. Chemistry 15:2036–2040
Bhunia A, Domadia PN, Torres J, Hallock KJ, Ramamoorthy A, Bhattacharjya S (2010) NMR structure of pardaxin, a pore-forming antimicrobial peptide, in lipopolysaccharide micelles: mechanism of outer membrane permeabilization. J Biol Chem 285:3883–3895
Bodey GP, Bolivar R, Fainstein V, Jadeja L (1983) Infections caused by Pseudomonas aeruginosa. Rev Infect Dis 5:279–313
Breidenstein EB, de la Fuente-Nunez C, Hancock RE (2011) Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol 19:419–426
Chen T, Farragher S, Bjourson AJ, Orr DF, Rao P, Shaw C (2003) Granular gland transcriptomes in stimulated amphibian skin secretions. Biochem J 371:125–130
Choi KY, Chow LN, Mookherjee N (2012) Cationic host defence peptides: multifaceted role in immune modulation and inflammation. J Innate Immun 4:361–370
Conlon JM (2011) Structural diversity and species distribution of host-defense peptides in frog skin secretions. Cell Mol Life Sci 68:2303–2315
Dathe M, Wieprecht T, Nikolenko H, Handel L, Maloy WL, MacDonald DL, Beyermann M, Bienert M (1997) Hydrophobicity, hydrophobic moment and angle subtended by charged residues modulate antibacterial and haemolytic activity of amphipathic helical peptides. FEBS Lett 403:208–212
Dathe M, Schumann M, Wieprecht T, Winkler A, Beyermann M, Krause E, Matsuzaki K, Murase O, Bienert M (1996) Peptide helicity and membrane surface charge modulate the balance of electrostatic and hydrophobic interactions with lipid bilayers and biological membranes. Biochemistry 35:12612–12622
De Zotti M, Biondi B, Formaggio F, Toniolo C, Stella L, Park Y, Hahm KS (2009) Trichogin GA IV: an antibacterial and protease-resistant peptide. J Pept Sci 15:615–619
De Zotti M, Biondi B, Park Y, Hahm KS, Crisma M, Toniolo C, Formaggio F (2012) Antimicrobial lipopeptaibol trichogin GA IV: role of the three Aib residues on conformation and bioactivity. Amino Acids 43:1761–1777
Di Grazia A, Cappiello F, Imanishi A, Mastrofrancesco A, Picardo M, Paus R, Mangoni ML (2015a) The frog skin-derived antimicrobial peptide esculentin-1a(1–21)NH2 promotes the migration of human HaCaT keratinocytes in an EGF receptor-dependent manner: a novel promoter of human skin wound healing? PLoS One 10:e0128663
Di Grazia A, Cappiello F, Cohen H, Casciaro B, Luca V, Pini A, Di YP, Shai Y, Mangoni ML (2015b) D-Amino acids incorporation in the frog skin-derived peptide esculentin-1a(1–21)NH2 is beneficial for its multiple functions. Amino Acids 47:2505–2519
Domadia PN, Bhunia A, Ramamoorthy A, Bhattacharjya S (2010) Structure, interactions, and antibacterial activities of MSI-594 derived mutant peptide MSI-594F5A in lipopolysaccharide micelles: role of the helical hairpin conformation in outer-membrane permeabilization. J Am Chem Soc 132:18417–18428
Drenkard E, Ausubel FM (2002) Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature 416:740–743
Epand RF, Savage PB, Epand RM (2007) Bacterial lipid composition and the antimicrobial efficacy of cationic steroid compounds (Ceragenins). Biochim Biophys Acta 1768:2500–2509
Epand RM, Vogel HJ (1999) Diversity of antimicrobial peptides and their mechanisms of action. Biochim Biophys Acta 1462:11–28
Erspamer V (1971) Biogenic amines and active polypeptides of the amphibian skin. Annu Rev Pharmacol 11:327–350
Free SJ (2013) Fungal cell wall organization and biosynthesis. Adv Genet 81:33–82
Gamberi T, Cavalieri D, Magherini F, Mangoni ML, De Filippo C, Borro M, Gentile G, Simmaco M, Modesti A (2007) An integrated analysis of the effects of Esculentin 1–21 on Saccharomyces cerevisiae. Biochim Biophys Acta 1774:688–700
Ganz T, Lehrer RI (1998) Antimicrobial peptides of vertebrates. Curr Opin Immunol 10:41–44
Gazit E, Lee WJ, Brey PT, Shai Y (1994) Mode of action of the antibacterial cecropin B2: a spectrofluorometric study. Biochemistry 33:10681–10692
Ghosh A, Bera S, Shai Y, Mangoni ML, Bhunia A (2016) NMR structure and binding of esculentin-1a (1–21)NH2 and its diastereomer to lipopolysaccharide: correlation with biological functions. Biochim Biophys Acta 1858:800–812
Giangaspero A, Sandri L, Tossi A (2001) Amphipathic alpha helical antimicrobial peptides. Eur J Biochem 268:5589–5600
Glukhov E, Stark M, Burrows LL, Deber CM (2005) Basis for selectivity of cationic antimicrobial peptides for bacterial versus mammalian membranes. J Biol Chem 280:33960–33967
Godwin U, Akpan MGS (2014) Effects of different concentrations of biocides on biofilm microorganisms in oil pipelines in Irri, Delta State, Nigeria. Int J Biosci 4:16
Gonzalez-Navajas JM, Corr MP, Raz E (2014) The immediate protective response to microbial challenge. Eur J Immunol 44:2536–2549
Griesinger COG, Wüthrich K, Ernst RR (1988) Clean TOCSY for proton spin system identification in macromolecules. J Am Chem Soc 110:7870–7872
Hancock RE, Rozek A (2002) Role of membranes in the activities of antimicrobial cationic peptides. FEMS Microbiol Lett 206:143–149
Hancock RE, Haney EF, Gill EE (2016) The immunology of host defence peptides: beyond antimicrobial activity. Nat Rev Immunol 16:321–334
Haney EF, Nathoo S, Vogel HJ, Prenner EJ (2010) Induction of non-lamellar lipid phases by antimicrobial peptides: a potential link to mode of action. Chem Phys Lipids 163:82–93
Haslam IS, Roubos EW, Mangoni ML, Yoshizato K, Vaudry H, Kloepper JE, Pattwell DM, Maderson PF, Paus R (2014) From frog integument to human skin: dermatological perspectives from frog skin biology. Biol Rev Camb Philos Soc 89:618–655
Hemshekhar M, Anaparti V, Mookherjee N (2016) Functions of cationic host defense peptides in immunity. Pharmaceuticals 9(3)
Hoiby N, Ciofu O, Johansen HK, Song ZJ, Moser C, Jensen PO, Molin S, Givskov M, Tolker-Nielsen T, Bjarnsholt T (2011) The clinical impact of bacterial biofilms. Int J Oral Sci 3:55–65
Islas-Rodriguez AE, Marcellini L, Orioni B, Barra D, Stella L, Mangoni ML (2009) Esculentin 1–21: a linear antimicrobial peptide from frog skin with inhibitory effect on bovine mastitis-causing bacteria. J Pept Sci 15:607–614
Karle IL, Balaram P (1990) Structural characteristics of alpha-helical peptide molecules containing Aib residues. Biochemistry 29:6747–6756
Kolar SS, Luca V, Baidouri H, Mannino G, McDermott AM, Mangoni ML (2015) Esculentin-1a(1–21)NH: a frog skin-derived peptide for microbial keratitis. Cell Mol Life Sci 72:617–627
Konig E, Bininda-Emonds OR, Shaw C (2014) The diversity and evolution of anuran skin peptides. Peptides 63C:96–117
Lee YR, Houngue C, Hall RG (2015) Treatment of community-acquired pneumonia. Expert Rev Anti Infect Ther 13:1109–1121
Levy SB (2002) The 2000 Garrod lecture. Factors impacting on the problem of antibiotic resistance. J Antimicrob Chemother 49:25–30
Lohner K (2016) Novel antibiotics based upon the multiple mechanisms of membrane perturbation by antimicrobial peptides. Curr Top Med Chem (in press)
Lohner K, Blondelle SE (2005) Molecular mechanisms of membrane perturbation by antimicrobial peptides and the use of biophysical studies in the design of novel peptide antibiotics. Comb Chem High Throughput Screen 8:241–256
Luca V, Stringaro A, Colone M, Pini A, Mangoni ML (2013) Esculentin(1–21), an amphibian skin membrane-active peptide with potent activity on both planktonic and biofilm cells of the bacterial pathogen Pseudomonas aeruginosa. Cell Mol Life Sci 70:2773–2786
Mangoni ML (2006) Temporins, anti-infective peptides with expanding properties. Cell Mol Life Sci 63:1060–1069
Mangoni ML, Shai Y (2011) Short native antimicrobial peptides and engineered ultrashort lipopeptides: similarities and differences in cell specificities and modes of action. Cell Mol Life Sci 68:2267–2280
Mangoni ML, Marcellini HG, Simmaco M (2007) Biological characterization and modes of action of temporins and bombinins H, multiple forms of short and mildly cationic anti-microbial peptides from amphibian skin. J Pept Sci 13:603–613
Mangoni ML, Luca V, McDermott AM (2015) Fighting microbial infections: A lesson from amphibian skin-derived esculentin-1 peptides. Peptides 71:286–295
Mangoni ML, Miele R, Renda TG, Barra D, Simmaco M (2001) The synthesis of antimicrobial peptides in the skin of Rana esculenta is stimulated by microorganisms. FASEB J 15:1431–1432
Mangoni ML, Fiocco D, Mignogna G, Barra D, Simmaco M (2003) Functional characterisation of the 1-18 fragment of esculentin-1b, an antimicrobial peptide from Rana esculenta. Peptides 24:1771–1777
Manning MC, Woody RW (1991) Theoretical CD studies of polypeptide helices: examination of important electronic and geometric factors. Biopolymers 31:569–586
Mansour SC, Pena OM, Hancock RE (2014) Host defense peptides: front-line immunomodulators. Trends Immunol 35:443–450
Matsuzaki K (2009) Control of cell selectivity of antimicrobial peptides. Biochim Biophys Acta 1788:1687–1692
Mookherjee N, Hancock RE (2007) Cationic host defence peptides: innate immune regulatory peptides as a novel approach for treating infections. Cell Mol Life Sci 64:922–933
Nicolas P, Mor A (1995) Peptides as weapons against microorganisms in the chemical defense system of vertebrates. Annu Rev Microbiol 49:277–304
Paiva AD, de Oliveira MD, de Paula SO, Baracat-Pereira MC, Breukink E, Mantovani HC (2012) Toxicity of bovicin HC5 against mammalian cell lines and the role of cholesterol in bacteriocin activity. Microbiology 158:2851–2858
Parsek MR, Tolker-Nielsen T (2008) Pattern formation in Pseudomonas aeruginosa biofilms. Curr Opin Microbiol 11:560–566
Ponti D, Mangoni ML, Mignogna G, Simmaco M, Barra D (2003) An amphibian antimicrobial peptide variant expressed in Nicotiana tabacum confers resistance to phytopathogens. Biochem J 370:121–127
Pouny Y, Rapaport D, Mor A, Nicolas P, Shai Y (1992) Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry 31:12416–12423
Rance M, Sorensen OW, Bodenhausen G, Wagner G, Ernst RR, Wuthrich K (1983) Improved spectral resolution in cosy 1H NMR spectra of proteins via double quantum filtering. Biochem Biophys Res Commun 117:479–485
Rink R, Arkema-Meter A, Baudoin I, Post E, Kuipers A, Nelemans SA, Akanbi MH, Moll GN (2010) To protect peptide pharmaceuticals against peptidases. J Pharmacol Toxicol Methods 61:210–218
Rybtke M, Hultqvist LD, Givskov M, Tolker-Nielsen T (2015) Pseudomonas aeruginosa Biofilm Infections: Community Structure, Antimicrobial Tolerance and Immune Response. J Mol Biol 427:3628–3645
Savjani JK, Gajjar AK, Savjani KT (2009) Mechanisms of resistance: useful tool to design antibacterial agents for drug—resistant bacteria. Mini Rev Med Chem 9:194–205
Schaffer C, Messner P (2005) The structure of secondary cell wall polymers: how Gram-positive bacteria stick their cell walls together. Microbiology 151:643–651
Schwermer CU, Lavik G, Abed RM, Dunsmore B, Ferdelman TG, Stoodley P, Gieseke A, de Beer D (2008) Impact of nitrate on the structure and function of bacterial biofilm communities in pipelines used for injection of seawater into oil fields. Appl Environ Microbiol 74:2841–2851
Shai Y (2002) Mode of action of membrane active antimicrobial peptides. Biopolymers 66:236–248
Shai Y, Oren Z (1996) Diastereoisomers of cytolysins, a novel class of potent antibacterial peptides. J Biol Chem 271:7305–7308
Simmaco M, Mignogna G, Barra D, Bossa F (1994) Antimicrobial peptides from skin secretions of Rana esculenta. Molecular cloning of cDNAs encoding esculentin and brevinins and isolation of new active peptides. J Biol Chem 269:11956–11961
Soufi Y, Soufi B (2016) Mass spectrometry-based bacterial proteomics: focus on dermatologic microbial pathogens. Front Microbiol 7:181
Strahilevitz J, Mor A, Nicolas P, Shai Y (1994) Spectrum of antimicrobial activity and assembly of dermaseptin-b and its precursor form in phospholipid membranes. Biochemistry 33:10951–10960
Toniolo C, Crisma M, Formaggio F, Peggion C (2001) Control of peptide conformation by the Thorpe-Ingold effect (C alpha-tetrasubstitution). Biopolymers 60:396–419
Toniolo C, Peggion C, Crisma M, Formaggio F, Shui X, Eggleston DS (1994) Structure determination of racemic trichogin A IV using centrosymmetric crystals. Nat Struct Biol 1:908–914
Toniolo C, Brückner H (2009) Peptaibiotics: fungal peptides containing α-dialkyl α-amino Acids. Wiley-VCH and VHCA, Weinheim/Zürich. Chem Biochem 10:2266–2267
Uccelletti D, Zanni E, Marcellini L, Palleschi C, Barra D, Mangoni ML (2010) Anti-Pseudomonas activity of frog skin antimicrobial peptides in a Caenorhabditis elegans infection model: a plausible mode of action in vitro and in vivo. Antimicrob Agents Chemother 54:3853–3860
Valenti P, Visca P, Antonini G, Orsi N (1985) Antifungal activity of ovotransferrin towards genus Candida. Mycopathologia 89:169–175
Veber DF, Freidinger RM (1985) The design of metabolically-stable peptide analogs. Trends Neurosci 8:392–396
Wüthrich K (1986) NMR of proteins and nucleic acids. Wiley, New York
Yamaguchi H, Kodama H, Osada S, Kato F, Jelokhani-Niaraki M, Kondo M (2003) Effect of alpha, alpha-dialkyl amino acids on the protease resistance of peptides. Biosci Biotechnol Biochem 67:2269–2272
Acknowledgments
This work was supported by grants from Sapienza Università di Roma and by FILAS Grant Prot. FILAS-RU-2014-1020. This article does not contain any studies with human participants or animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling Editor: M. S. Palma.
Rights and permissions
About this article

Cite this article
Biondi, B., Casciaro, B., Di Grazia, A. et al. Effects of Aib residues insertion on the structural–functional properties of the frog skin-derived peptide esculentin-1a(1–21)NH2 . Amino Acids 49, 139–150 (2017). https://doi.org/10.1007/s00726-016-2341-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-016-2341-x