
Abstract
Several experimental approaches have demonstrated that transglutaminase 2 (TG2) increased activity is involved in monocyte activation and inflammatory response. Preliminary results also demonstrate a TG-mediated post-translational modification of phospholipase A2 (PLA2), which catalyzes the release of arachidonic acid from its lipid storage sites. The control of PLA2-mediated production of eicosanoids has been found to be of great benefit for inflammatory disease treatment. However, the identification of the mechanisms of PLA2 activation is a very complex issue, because of the presence of multiple PLA2 forms. The aim of this study was to characterize the interactions between TG2 and sPLA2 in LPS-stimulated THP-1 cells, which were treated with TPA to induce early differentiated macrophage-type model. We demonstrated that increases in TG2 enzyme activity and protein expression may be considered an early event in monocyte/macrophage activation by LPS. Under these conditions, TG2 protein was co-immunoprecipitated with PLA2 by monoclonal antibody directed against the secretory form of the enzyme (sPLA2-V). Concomitantly, the PLA2 enzyme activity increased in TPA-treated cells exposed to LPS; these high levels of enzyme activity were significant reduced by R283, a site-specific inhibitor of TG2. Moreover, confocal laser scanning microscopy analysis of double-immunostained cytochemical specimens confirmed a co-localization of BAPA-labeled proteins and sPLA2-V in LPS-treated cells. These findings give evidence of a complex TG2/sPLA2-V, suggesting the possibility that sPLA2-V is a substrate for TG2. These results demonstrated that TG2 increases produced a sustained activation of PLA2 activity, suggesting a functional interaction between these enzymes in the regulation of inflammatory response.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Transglutaminase 2 (TG2) is the most ubiquitous member of transglutaminase family enzymes, which catalyze calcium-dependent post-translational protein modifications through either the formation of ε(γ-glutamyl)lysine cross-links between glutamine and lysine residues or incorporation of primary amines and polyamines (Aeschlimann and Paulsson 1994; Griffin et al. 2002). TG2 may also bind and hydrolyze GTP and ATP, acting as a signal transduction G protein, and even display other enzyme activities, catalyzing either protein disulfide isomerase or protein kinase reactions (Cervellati et al. 2012; Griffin et al. 2002; Park et al. 2010).
TG2 is a multifunctional protein which may be secreted to the cell surface by an unknown mechanism (Wang and Griffin 2012). TG2 is constitutively expressed in many different cell types, and depending on the cell type can be regulated by several transcriptional activators, such as cytokines, retinoids, vitamin D and steroid hormones (Krig et al. 2002; Quan et al. 2005; Garabuczi et al. 2013). The aberrant induction of TG2 activity contributes to various pathologies, including neurodegenerative diseases, atherosclerosis, autoimmune diseases and fibrosis (Kim et al. 2002; Szondy et al. 2011). More recently, several experimental approaches have demonstrated that TG2 activity increased both in diseased tissue with inflammation and in cells with inflammatory stress (Kim 2006). Under these conditions, a specific cross-talk between TG2 and nuclear transcription factor-κB (NF-κB) appeared to be active. Indeed, TGM2 gene transcription can be directly induced by NF-κB activation, because the TGM2 promoter has an NF-κB binding motif (Mirza et al. 1997) and, conversely, an increase in TG activity is associated with alternative pathways of NF-κB activation (Ientile et al. 2007; Kumar and Mehta 2012).
The activation of macrophages is a critical event in the inflammatory response and several results demonstrate that increases in TG2 enzyme activity are involved in macrophage activation (Novogrodsky et al. 1978; Schroff et al. 1981; Murtaugh et al. 1983).
It has long been known that TG2 non-covalently interacts in vitro with various proteins such as those involved in ECM formation and stabilization (Griffin et al. 2002; Turner and Lorand 1989). However, due to the different properties of TG2 enzyme and its involvement in various pathways, it is not possible to exclude that other substrates can be dependent on TG2-induced post-translational modification in activated macrophages.
A preliminary study demonstrated a TG-mediated post-translational modification of porcine pancreatic phospholipase A2 (PLA2), leading to a dramatic activation of this enzyme (Cordella-Miele et al. 1990). The main role of PLA2 enzyme family is associated with pathogenesis of inflammation through hydrolysis of arachidonic acid (AA) from phospholipids (Chakraborti 2003). Actually, there are several phospholipases A2 that comprise a family of different enzymes identified through their nucleotide gene sequences, and classified into three main groups: (i) cytosolic PLA2 (cPLA2), (ii) secretory PLA2 (sPLA2), and (iii) intracellular PLA2 (iPLA2). They differ from each other in terms of substrate specificity, Ca2+ requirement and lipid modification.
Studies on the cPLA2 knock-out mouse and derived cells have demonstrated the importance of this enzyme in a variety of physiological and pathophysiological states. The chronic exposure of macrophages to LPS results in sPLA2 activation stimulating iNOS expression and nitric oxide production by NF-κB dependent mechanism (Baek et al. 1950).
Previous studies have demonstrated the TG2-catalyzed post-translational modification of PLA2. In fact, when PLA2 was incubated with TG2 in the presence of putrescine, spermine, spermidine, dansylcadaverine, or methylamine, a two-to-threefold increase in PLA2 activity was observed (Cordella-Miele et al. 1993).
Since TG2 and sPLA2 activation has the potential to bring about relevant changes in cellular metabolism in response to inflammatory stimuli, the aim of this study was to characterize in LPS-stimulated THP-1 cells the interactions between TG2 and sPLA2 group V (sPLA2-V), a member of the superfamily of PLA2 enzymes characterized by the ability to hydrolyze the sn-2 ester bond of phospholipids and cell membranes.
Materials and methods
Materials
The human pre-monocytic cell line, THP-1, was obtained from DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen-Braunschweig, Germany).
RPMI-1640, penicillin/streptomycin mixture, l-glutamine, HEPES, sodium pyruvate, glucose, aprotinin, leupeptin, pepstatin, 12-O-tetradecanoylphorbol-13-acetate (TPA), calcium ionophore A23187 and other chemicals of analytical grade were from Sigma (Milan, Italy). Fetal bovine serum (FBS), M-SFM, EnzChek® Phospholipase A2 Assay Kit as well as TRIzol for RNA extraction were from Invitrogen Life Technologies (Milan, Italy). LPS was purchased from Invivogen (San Diego, California, USA). 5-(Biotinamido)pentylamine (BAPA) was from Pierce Biotechnology Inc. (Rockford, IL). Monoclonal antibody for TG2 was from Neomarkers (Fremont, CA). Monoclonal antibody for sPLA2-V was from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal antibody for β-actin, horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody, and streptavidin-FITC were from Sigma (Milan, Italy). ECL Chemiluminescence detection kit and X-ray film were from Amersham Pharmacia Biotech (Milan, Italy). Developer, fixer and Kodak X-ray film were from Kodak (Milan, Italy).
High-capacity cDNA archive kit, TaqMan Gene Expression Mastermix, TaqMan Gene Expression assays (Assays-on-Demand) for human β-actin (ID: Hs99999903_m1) and TGM2 (ID: Hs00190278_m1) were from Applied Biosystems (Life Technologies, Milan, Italy).
1,3-Dimethyl-2[(oxopropyl)thio]imidazolium (R283) was a generous gift from Professor Martin Griffin (Aston University, Birmingham, UK).
Cell culture and treatment
THP-1 cells were maintained in RPMI 1640 supplemented with l-glutamine (2 mM), HEPES (10 mM), sodium pyruvate (1 mM), glucose (2.5 g/l), and 10 % heat-inactivated fetal bovine serum, at 37 °C in a 5 % CO2/95 % air humidified atmosphere. The medium was renewed every 2 days and split was performed when cells reached maximum density (1 × 106 cells/ml).
To induce monocyte differentiation to macrophages, THP-1 cells were seeded in six-well culture plates at a density of 5 × 105 cells/ml in M-SFM with 200 nM TPA and incubated for 24 h.
After differentiation, the cells were washed twice with PBS and treated with or without either LPS (0.01–0.5 μg/ml) or TNF-α (0.1–5 ng/ml) in fresh RPMI with 10 % heat-inactivated FBS for 4–24 h.
In parallel experiments, TG2 activation was achieved by addition of calcium ionophore A23187 (100 μM) to the cultures 2 h prior to the end of the incubation period.
For TG2 inhibition, the site-directed TG2 inhibitor R283 (250 μM) was added to the culture medium 2 h before LPS exposure.
Analysis of mRNA expression by real-time PCR
Total RNA was isolated from stimulated and non-stimulated cells using TRIzol reagent (Invitrogen, Milan, Italy). Two micrograms of RNA were reverse transcribed with High-Capacity cDNA Archive kit according to the manufacturer’s instructions. Then, TGM2 mRNA levels was analyzed by real-time PCR using a TaqMan gene expression assay (Life Technologies, Milan, Italy). β-Actin was used as endogenous control. Quantitative PCR reactions were set up in duplicate in a 96-well plate and carried out in 20 μl reactions containing 1× Gene Expression Mastermix, 1× TaqMan-specific assay, and 25 ng RNA converted into cDNA. Real-time PCR was performed in a 7900HT Fast Real-Time PCR System with the following profile: one cycle at 50 °C for 2 min, then 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min. Data were collected and analyzed using SDS 2.3 and RQ manager 1.2 software (Applied Biosystems, Foster City, CA) using the 2−ΔΔCT relative quantification method. Values are presented as fold change relative to unstimulated cells.
Western blotting
To obtain whole cell extracts, cells were lysed using ice-cold RIPA buffer supplemented with protease inhibitor cocktail (SIGMA Aldrich, Milan, Italy) and cell debris were removed by centrifugation at 8,000×g at 4 °C for 20 min. Protein concentration was evaluated by the Bradford method and 30 µg of total protein was loaded on a 10 % denaturing SDS–polyacrylamide gel, and transferred to nitrocellulose membranes. After protein transfer, the membranes were blocked with Tris-buffered saline Tween-20 (TBS-T) with 5 % non-fat dry milk at room temperature for an hour. Detection of specific proteins was done by probing membranes with mouse monoclonal antibodies against TG2 (CUB7402, diluted 1:1,000 in TBS-T) and sPLA2-V (diluted 1:500 in TBS-T) for 2 h at room temperature, followed by incubation with horseradish peroxidase-conjugated anti-mouse (diluted 1:2,000 in TBS-T) for 2 h at room temperature. Immunoblots were developed with ECL Plus chemiluminescent detection system kit using Kodak film. The bands were scanned and quantified by densitometric analysis with an AlphaImager 1200 System (Alpha Innotech, San Leandro, CA, USA), after normalization against β-actin.
In situ TG activity assay
To measure in situ TG enzymatic activity, the incorporation of 5-(biotinamido)pentylamine (BAPA) into proteins was quantified by using a colorimetric assay as previously described (Zhang et al. 1998). In brief, 1 h prior to the end of incubation period, 1 mM 5-(biotinamido)pentylamine (BAPA) was added to the culture medium. Then, cells were homogenized, sonicated on ice, and protein concentration was determined. Ten micrograms of protein was diluted with coating buffer, loaded into each well of a 96-well microtiter plate and incubated overnight at 4 °C. After blocking non-specific binding sites, 100 µl of HRP-conjugated streptavidin (1:1,000) in 1 % BSA and 0.01 % Tween 20 in borate saline buffer was added to each well and incubated at room temperature for 1 h.
After washing, 200 μl of substrate solution (0.4 mg of o-phenylenediamine dihydrochloride/ml of 0.05 M sodium citrate phosphate buffer, pH 5.0) was added to each well. The reactions were stopped by 3 N HCl, and the presence of biotinylated proteins was quantified by measuring the absorbance at 492 nm on a microplate spectrophotometer (Tecan). All measurements were done in triplicate and repeated at least three times.
Measurement of PLA2 activity
PLA2 activity measurement was performed using the EnzChek® Phospholipase A2 Assay Kit (Invitrogen) as recommended by the manufacturer. After stimulation, cells were washed, harvested with a nonenzymatic dissociation solution and lysed in homogenization buffer (50 mM Tris, pH 7.5, 150 mM NaCl with protease inhibitor cocktail).
Cell lysates (60 μg) were incubated with substrate–liposome mix in a 96-well plate for 10 min at room temperature in the dark, and then the fluorescence was measured at Ex/Em 460/515 nm using a microplate reader (Tecan, Milan, Italy).
Confocal laser scanning microscopy (CLSM)
The co-localization of sPLA2-V protein and TG enzymatic activity was examined in control and exposed cells plated at a density of 2.5 × 104 cells/well onto 4-well chamber slide, on a Leica confocal laser scanning microscope (Laborlux K, Leica Microsystems GmbH Heidelberg, Mannheim, Germany).
One hour prior to the end of the incubation period, 1 mM BAPA was added to the culture medium. At the end of each treatment, cells were fixed with a paraformaldehyde solution (4 %) for 15 min and permeabilized with 0.1 % Triton X-100 in PBS for 2 min, before incubation with antibody against sPLA2-V (1/50 in PBS) for 2 h at room temperature. After three washes with PBS, slides were incubated 1 h at room temperature with streptavidin-FITC (1:100 in PBS) for BAPA and TRITC-labeled anti-mouse IgG (1:64 in PBS) for sPLA2-V. Finally, slides were washed with PBS, mounted with 90 % glycerol and observed with CLSM.
Immunoprecipitation
To detect the interaction between TG2 and sPLA2-V, cells were lysed in a Tris–HCl buffer containing 20 mM Tris–HCl (pH 7.5) and protease inhibitors. Protein concentration was evaluated by the Bradford method and equal amounts of proteins (500 μg) were incubated with sPLA2 monoclonal antibody for 2 h at 4 °C. Immunocomplexes were incubated with Protein G Plus-Agarose beads (Invitrogen) overnight at 4 °C, then washed, and boiled for 5 min in 1×SDS protein loading dye. Negative control was set by incubating cell lysates under similar conditions, but without the immunoprecipitating antibody. Samples were loaded on a 7.5 % denaturing SDS–polyacrylamide gel, and transferred to nitrocellulose membranes. After blocking, membranes were probed with antibody against TG2 (CUB 7402, diluted 1:1,000 in TBS-T) and then with horseradish peroxidase-conjugated anti-mouse secondary antibody (diluted 1:2,000 in TBS-T) for 2 h at room temperature. Detection was performed using ECL Plus chemiluminescent detection system.
Statistical analysis
All experiments were repeated at least three times and each experiment was performed at least in duplicate. All values are expressed as mean ± standard error of the mean (SEM). Statistical analysis was carried out using Student’s t test for comparisons between the two groups, with p values less than 0.05 considered significant.
Results
In this study, we used the human monocytic THP-1 leukemia cell line that has been widely accepted as a good model of monocytes/macrophages (Qin 2012). THP-1 monocytes were treated with TPA to induce early differentiation toward macrophage phenotype and subsequently treated for different times (4–24 h) with or without either LPS or TNF-α, as pro-inflammatory agents. First, we investigated whether TG2 can be directly involved in THP-1 cell activation induced by LPS or TNF-α. Preliminary results from these experiments demonstrated that in TPA-differentiated THP-1 cells, TGM2 transcription was not affected by 4 h treatment with either LPS (0.01–0.5 μg/ml) or TNF-α (0.1–5 ng/ml), while dose-dependent increases were observed after 24 h in presence of both LPS or TNF-α. In particular, the most effective doses resulted in 0.5 μg/ml for LPS and 5 ng/ml for TNF-α that produced a fourfold and threefold increase, respectively, in TG2 mRNA transcript levels in comparison to controls (Fig. 1). Higher doses showed beside toxic effects without further increasing mRNA transcript levels (data not shown).

Analysis of TG2 expression in TPA-differentiated THP-1 cells after stimulation with LPS (0.5 μg/ml) or TNF-α (5 ng/ml) for 4–24 h. TG2 mRNA levels were evaluated by quantitative real-time PCR, after normalization against β-actin as endogenous control, as described in “Materials and methods”. The data represent the mean ± SEM from three separate experiments. *p< 0.05 significant differences in comparison with control
Given the higher TG2 induction achieved by LPS treatment compared with TNF-α, subsequent experiments were carried out by using 0.5 μg/ml LPS as inflammatory stimulus.
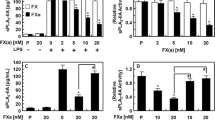
LPS-induced TG2 upregulation, in TPA-differentiated THP-1 cells, was confirmed by Western blot analysis that showed a twofold TG2 protein increase in 24 h LPS-treated cells in comparison to untreated ones (Fig. 2a). Under our experimental conditions, the exposure to 0.5 μg/ml LPS also caused slight increases in the expression of sPLA2-V. In particular, a 24 h THP1 cell incubation with LPS produced a 1.5 fold increase of sPLA2-V.

Effects of LPS stimulation on TG2 and PLA2 expression in differentiated THP-1 cells. Western blot analysis of TG2 (a) and sPLA2 (b). Results of densitometric analysis, after normalization against β-actin levels, are mean ± SEM from three separate experiments. *p< 0.05 significant differences in comparison to control cells
It has been postulated that in the inflammatory cells, sPLA2 and TG2 can interact and further PLA2 activation may occur as this process continues (Moreno 2006).
The intracellular TG-catalyzed reaction products can be detected by evaluating BAPA incorporation into cell proteins. Given that TG2 transamidating activity is dependent on calcium availability, we evaluated the incorporation of BAPA (1 mM) in TPA-differentiated THP-1 cells after stimulation with LPS (0.5 μg/ml) in the presence or absence of the calcium ionophore A23187 (100 μM). A 24 h exposure to LPS increased by 80 % (p < 0.05) TG2 activity in comparison to controls. This increase was similar to that achieved, at the same time, in the presence of the calcium ionophore A23187 (Fig. 3a). Moreover, a slight increase was observed in THP-1 cells incubated with LPS and A23187 (Fig. 3a).

Effect of LPS stimulation on TG and PLA2 activity of differentiated THP-1 cells. a TG-mediated BAPA incorporation in THP-1 cell cultures exposed to LPS (0.5 μg/ml) for 4–24 h in the presence or absence of calcium ionophore A23187 or R283. In situ TG activity was determined as described in “Materials and methods”. Data are mean ± SEM from three independent determinations. * and § p < 0.05 significant differences in comparison to control and LPS, respectively. b PLA2 activity was determined in control and LPS-stimulated THP-1 cells incubated either in the absence or presence of R283, the TG2 site-specific inhibitor, as described in “Materials and methods”. Data are mean ± SEM from three separate experiments. *p< 0.05 and § p< 0.05 significant differences in comparison to control and LPS, respectively
In parallel, the PLA2 enzyme activity increased in TPA-treated cells exposed to LPS (0.5 μg/ml), reaching about 60 % increase in comparison to untreated cells (Fig. 3b). We also evaluated the effect of TG2 inhibition on PLA2 enzyme activity. The pre-incubation with R283, the site-specific inhibitor of TG2, while significantly reducing LPS-increased TG2 activity also produced a decrease of PLA2 activity (Fig. 3b).
Confocal laser scanning microscopy analysis demonstrated that LPS-treated cells showed an intense fluorescent signal of BAPA labeling, due to TG-mediated incorporation into cell proteins, in cytosol and perinuclear compartment (Fig. 4a). This effect was also evident by intensities of fluorescence measured in LPS-treated cells in comparison to controls (Fig. 4b).

a Confocal laser scanning micrographs showing the co-localization of TG-mediated BAPA labeling of protein substrates and sPLA2 in differentiated THP-1 cell cultures exposed to LPS (0.5 μg/ml) for 24 h. BAPA (1 mM) was added to the culture medium 1 h prior to the end of incubation; then, cells were incubated with anti-sPLA2 followed by TRITC-labeled anti-mouse IgG and FITC-streptavidin as described in “Materials and methods”. TG activity is in green, and PLA2 localization is in red. Merging of the images shows co-localization of transglutaminase activity and sPLA2 (yellow). b The results of mean fluorescence intensity are reported. The fluorescent signal intensity from both BAPA labeled structures and sPLA2-V in LPS-treated cells were higher than the respective controls (color figure online)
Unstimulated cells also revealed a diffuse pattern of cytoplasmic staining with anti-sPLA2-V antibody (Fig. 4a). When LPS-treated cells were challenged with antibody against sPLA2-V, a marked fluorescence overlapping with BAPA signaling was evident. Merged images showing the co-localization of cytoplasmic BAPA incorporation and PLA2 immunofluorescent labeling in THP-1 cells in the presence of LPS are shown in Fig. 4a.
In addition, to investigate the possible interaction between these enzymes in TPA-differentiated cells upon LPS incubation, co-immunoprecipitation experiments were carried out by using sPLA2-V monoclonal antibody and TG2-specific antibody. The results obtained showed that TG2 co-immunoprecipitated with sPLA2-V both in untreated and LPS-treated cells, suggesting that TG2 forms a complex with sPLA2 in THP-1 cells. However, the intensity of TG2 immunoreactive band was increased in LPS-treated cells (Fig. 5).

TG2 co-immunoprecipitates with sPLA2 in differentiated THP-1 cells. After 24 h of treatment with LPS (0.5 μg/ml), treated and relative control cells were lysed and proteins were immunoprecipitated using anti-sPLA2 antibody. Immunoprecipitated proteins were separated by SDS-PAGE and immunoblotted for TG2. IN input fraction, IP immunoprecipitate, OUT supernatant after immunoprecipitation. CTR control cells, LPS LPS-treated cells
Discussion
Relatively few information is known about the underlying mechanisms following the increases in TG activity as a critical event in monocyte/macrophage activation and inflammatory response. Macrophages are involved in the inflammation or tissue injury, and they are active in cytokine production. Macrophages at these sites also synthesize large amounts of TG2 in response to different inflammatory stimuli including LPS (Mehta et al. 2010). The differentiation of monocytes into macrophages is associated with TG2-upregulation and a concomitant decrease in blood-clotting factor XIIIa levels (Seiving et al. 1991). Here, we report that increases in TG2 mRNA transcripts may be considered an early event in differentiated monocytes stimulated with LPS. In this regard, we recently also reported that TG2 might be required for the functional activation of monocytes by amyloid peptide, given that the expression of cell surface markers and adhesion molecules, such as CD14 and fibronectin, as well as pro-inflammatory mediators, such as TNF-α and MMP-9, were found to depend on amyloid peptide-induced TG2 upregulation (Currò et al. 2010).
We also demonstrated that TG2 enzyme activity, evaluated through primary amine BAPA incorporation into cell proteins, was increased in comparison to control cells after LPS treatment, reaching levels similar to those observed in the presence of calcium ionophore A23187.
Although intracellular calcium concentrations rarely increased to levels high enough (millimolar) to stimulate TG2 cross-linking activity, intracellular TG-catalyzed isopeptide bonds have been recently reported (Király et al. 2011; Verhaar et al. 2011). Additionally, many efforts were made to identify substrate proteins and interaction partners for transglutaminases and interactive database are increasing (http://genomics.dote.hu.wiki). Given the availability of cellular pools of polyamines, TG-mediated protein modifications by polyamination are likely to occur more frequently than cross-linking, since polyamination requires lower intracellular calcium levels than the threshold needed for cross-linking.
Our results using confocal laser scanning microscopy analysis gave evidence for a co-localization of BAPA-labeled substrates and sPLA2-V staining in LPS-treated cells, and we also demonstrated that both TG2 protein and sPLA2-V co-immunoprecipitated in activated macrophages. These results suggest that sPLA2 may act as a substrate for TG2 primary amine incorporation.
Interestingly, a previous study indicated that PLA2 polyamination may contribute to the inflammation associated with neurodegeneration (Jeitner et al. 2009).
The sPLA2s are all low-molecular mass proteins, requiring millimolar concentrations of Ca2+ for enzyme activity. The sPLA2 participates in a variety of pathological processes by releasing arachidonic acid from membrane phospholipids, leading to the production of various types of pro-inflammatory lipid mediators such as prostaglandins, thromboxanes and leukotrienes (Chakraborti 2003). Under our conditions, the incubation with calcium ionophore significantly increased TG2 enzyme activity, similarly to what was observed in the presence of LPS. Moreover, TG2 activity inhibition caused a significant reduction in LPS-increased PLA2 activity. This may be due to an interaction between transamidating TG activity and sPLA2-V in LPS-exposed cells.
On the basis of these results, the increase in BAPA incorporation suggest that polyamination may be a mechanism involved in the intracellular sPLA2-V translation toward the membrane, and/or to regulate protein turnover. Indeed, previous results demonstrate that polyamination of PLA2 results in a threefold increased activity (Cordella-Miele et al. 1993), and this post-translational protein modification may persist for the life of the protein given the inability of most peptidases to hydrolyze γ-glutamylamine (GGEL, γ-glutamylpolyamine and bis-γ-glutamylpolyamine) linkages (Fink and Folk 1981).
Previous results demonstrated that the anti-inflammatory effects of antinflammins may be, at least in part, due to the inhibition of TG activity (Moreno 2006). Therefore, it is possible to hypothesize that TG2 inhibitors, such as antinflammins, may have dual anti-inflammatory properties, decreasing cell infiltration and TG2-dependent PLA2 activation and these inhibitory effects may be able to reduce the inflammatory process.
To summarize, we show that TG2 increases evoked by inflammatory stimulus produced a sustained activation of sPLA2. Because PLA2 play a role in the first rate-limiting step leading to eicosanoid production, these findings may reveal a functional interaction between these enzymes leading to a mechanism useful to amplify inflammatory response.
References
Aeschlimann D, Paulsson M (1994) Transglutaminases: protein cross-linking enzymes in tissues and body fluids. Thromb Haemost 71:402–415
Baek SH, Kwon TK, Lim JH et al (2000) Secretory phospholipase A2-potentiated inducible nitric oxide synthase expression by macrophages requires NF-kappa B activation. J Immunol 164:6359–6365
Cervellati C, Montin K, Squerzanti M et al (2012) Effects of the regulatory ligands calcium and GTP on the thermal stability of tissue transglutaminase. Amino Acids 42:2233–2242. doi:10.1007/s00726-011-0963-6
Chakraborti S (2003) Phospholipase A(2) isoforms: a perspective. Cell Signal 15:637–665
Cordella-Miele E, Miele L, Mukherjee AB (1990) A novel transglutaminase-mediated post-translational modification of phospholipase A2 dramatically increases its catalytic activity. J Biol Chem 265:17180–17188
Cordella-Miele E, Miele L, Beninati S, Mukherjee AB (1993) Transglutaminase-catalyzed incorporation of polyamines into phospholipase A2. J Biochem 113:164–173
Currò M, Ferlazzo N, Condello S et al (2010) Transglutaminase 2 silencing reduced the beta-amyloid-effects on the activation of human THP-1 cells. Amino Acids 39:1427–1433. doi:10.1007/s00726-010-0605-4
Fink ML, Folk JE (1981) Gamma-glutamylamine cyclotransferase. An enzyme involved in the catabolism of epsilon-(gamma-glutamyl) lysine and other gamma-glutamylamines. Mol Cell Biochem 38:59–67
Garabuczi É, Kiss B, Felszeghy S et al (2013) Retinoids produced by macrophages engulfing apoptotic cells contribute to the appearance of transglutaminase 2 in apoptotic thymocytes. Amino Acids 44:235–244. doi:10.1007/s00726-011-1119-4
Griffin M, Casadio R, Bergamini CM (2002) Transglutaminases: nature’s biological glues. Biochem J 368:377–396. doi:10.1042/BJ20021234
Ientile R, Caccamo D, Griffin M (2007) Tissue transglutaminase and the stress response. Amino Acids 33:385–394. doi:10.1007/s00726-007-0517-0
Jeitner TM, Pinto JT, Krasnikov BF et al (2009) Transglutaminases and neurodegeneration. J Neurochem 109:160–166. doi:10.1111/j.1471-4159.2009.05843.x
Kim SY (2006) Transglutaminase 2 in inflammation. Front Biosci J Virtual Libr 11:3026–3035
Kim SY, Jeitner TM, Steinert PM (2002) Transglutaminases in disease. Neurochem Int 40:85–103
Király R, Demény M, Fésüs L (2011) Protein transamidation by transglutaminase 2 in cells: a disputed Ca2+-dependent action of a multifunctional protein. FEBS J 278:4717–4739. doi:10.1111/j.1742-4658.2011.08345.x
Krig SR, Chandraratna RAS, Chang MMJ et al (2002) Gene-specific TCDD suppression of RARalpha- and RXR-mediated induction of tissue transglutaminase. Toxicol Sci Off J Soc Toxicol 68:102–108
Kumar S, Mehta K (2012) Tissue transglutaminase constitutively activates HIF-1α promoter and nuclear factor-κB via a non-canonical pathway. PLoS ONE 7:e49321. doi:10.1371/journal.pone.0049321
Mehta K, Kumar A, Kim HI (2010) Transglutaminase 2: a multi-tasking protein in the complex circuitry of inflammation and cancer. Biochem Pharmacol 80:1921–1929. doi:10.1016/j.bcp.2010.06.029
Mirza A, Liu SL, Frizell E et al (1997) A role for tissue transglutaminase in hepatic injury and fibrogenesis, and its regulation by NF-kappaB. Am J Physiol 272:G281–G288
Moreno JJ (2006) Effects of antiflammins on transglutaminase and phospholipase A2 activation by transglutaminase. Int Immunopharmacol 6:300–303. doi:10.1016/j.intimp.2005.08.001
Murtaugh MP, Mehta K, Johnson J et al (1983) Induction of tissue transglutaminase in mouse peritoneal macrophages. J Biol Chem 258:11074–11081
Novogrodsky A, Quittner S, Rubin AL, Stenzel KH (1978) Transglutaminase activity in human lymphocytes: early activation by phytomitogens. Proc Natl Acad Sci U S A 75:1157–1161
Park D, Choi SS, Ha K-S (2010) Transglutaminase 2: a multi-functional protein in multiple subcellular compartments. Amino Acids 39:619–631. doi:10.1007/s00726-010-0500-z
Qin Z (2012) The use of THP-1 cells as a model for mimicking the function and regulation of monocytes and macrophages in the vasculature. Atherosclerosis 221:2–11. doi:10.1016/j.atherosclerosis.2011.09.003
Quan G, Choi J-Y, Lee D-S, Lee S-C (2005) TGF-beta1 up-regulates transglutaminase two and fibronectin in dermal fibroblasts: a possible mechanism for the stabilization of tissue inflammation. Arch Dermatol Res 297:84–90. doi:10.1007/s00403-005-0582-8
Schroff G, Neumann C, Sorg C (1981) Transglutaminase as a marker for subsets of murine macrophages. Eur J Immunol 11:637–642. doi:10.1002/eji.1830110809
Seiving B, Ohlsson K, Linder C, Stenberg P (1991) Transglutaminase differentiation during maturation of human blood monocytes to macrophages. Eur J Haematol 46:263–271
Szondy Z, Korponay-Szabó I, Király R, Fésüs L (2011) Transglutaminase 2 dysfunctions in the development of autoimmune disorders: celiac disease and TG2-/- mouse. Adv Enzymol Relat Areas Mol Biol 78:295–345
Turner PM, Lorand L (1989) Complexation of fibronectin with tissue transglutaminase. Biochemistry 28:628–635
Verhaar R, Jongenelen CAM, Gerard M et al (2011) Blockade of enzyme activity inhibits tissue transglutaminase-mediated transamidation of α-synuclein in a cellular model of Parkinson’s disease. Neurochem Int 58:785–793. doi:10.1016/j.neuint.2011.03.004
Wang Z, Griffin M (2012) TG2, a novel extracellular protein with multiple functions. Amino Acids 42:939–949. doi:10.1007/s00726-011-1008-x
Zhang J, Lesort M, Guttmann RP, Johnson GV (1998) Modulation of the in situ activity of tissue transglutaminase by calcium and GTP. J Biol Chem 273:2288–2295
Acknowledgments
We thank Prof. M. Griffin (Aston University, Birmingham, UK) for the gift of the TG2-specific inhibitor R283.
Conflict of interest
The authors disclose no potential conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Currò, M., Ferlazzo, N., Risitano, R. et al. Transglutaminase 2 and phospholipase A2 interactions in the inflammatory response in human Thp-1 monocytes. Amino Acids 46, 759–766 (2014). https://doi.org/10.1007/s00726-013-1569-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-013-1569-y