
Abstract
The well-defined unnatural dipeptides based on cis-2,5-disubstituted pyrrolidine backbone were synthesized from commercially available starting materials meso-diethyl-2,5-dibromoadipate, (S)-(−)-1-phenylethylamine, and phenylalanine. The configurations of all the chiral centers in these unnatural dipeptides are confirmed by X-ray crystal diffraction analysis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The unnatural peptides play a key role in peptidomimetics (Sun et al. 2008; Hanessian and Auzzas 2008; Isidro-Llobet et al. 2011; Ung and Winkler 2011), among them peptides containing 2,5-disubstituted pyrrolidine moiety have drawn much attention of chemists and pharmaceutists by their unique physiological activities (Wang et al. 2001; Enache et al. 2009; Davis et al. 2008; Draper and Britton 2010). Due to non-planar nature of pyrrolidine, peptides containing pyrrolidine motif can have flexible conformation which is responsible for their biological activities (Madis 1977; Giordano et al. 2010; Warren et al. 2010). Pei et al. (2006) have found a series of unnatural dipeptides based on cis-2,5-disubstituted pyrrolidine backbone which were used as potent dipeptidyl peptidase IV (DPP-IV) inhibitors for potential anti-diabetic drugs. Manfré and Pulicani (1994) synthesized a unnatural dipeptide (+)-RP-66803 with cis-2,5-disubstituted pyrrolidine core as a cholecystokinin (CCK) antagonist (Fig. 1). The compounds containing 2,5-disubstituted pyrrolidine ring are also attractive to synthetic chemists by their versatile utilities in organic synthesis (Vaswani et al. 2009; Lemen and Wolfe 2010; Chen et al. 2009; Shu et al. 2010). Some important alkaloids, such as cocaine (Lewin et al. 1987; Mans and Pearson 2004), gephyrotoxin (Miao et al. 2010) and monomorine (Jefford et al. 1991), possess cis-2,5-disubstituted pyrrolidine ring system. The synthesis of several indolizidine alkaloids containing a chiral trans-2,5-disubstituted pyrrolidine moiety was described (Dhimane et al. 1997; Takahata et al. 1990; Wang et al. 1999; Lee et al. 2000; Cahill et al. 1999); however, only few methods provide stereoselective synthesis of cis-2,5-disubstituted pyrrolidine derivatives (Haddad et al. 1998; Brenneman and Martin 2004; Bagley and Tovey 2001; Colandrea et al. 2006). Lygo et al. (2010) have described a synthetic route to cis-2,5-disubstituted pyrrolidine derivatives by asymmetric phase-transfer catalysis. Previously, we have reported a facile access to enantiopure cis-2,5-disubstituted pyrrolidines (Fig. 1, compounds A and B) using meso-diethyl-2,5-dibromoadipate and (S)-(−)-1-phenylethylamine as starting materials (Wang et al. 2007). With this background and in continuation of our previous work, we report in this paper an efficient construction of unnatural dipeptides from phenylalanine and cis-2,5-disubstituted pyrrolidines. In which configuration of the chiral centers was confirmed by X-ray crystallographic analysis.

Some novel unsymmetric cis-2,5-disubstituted pyrrolidines
Experimental
General
1H NMR and 13C NMR spectra were measured in CDCl3, MeOD or DMSO-D6 solution on a Bruker AV-300 or AV-500 spectrometer using TMS as an internal reference. Coupling constant (J) values are given in Hz. Multiplicities are designated by the following abbreviations: s, singlet; d, doublet; t, triplet; q, quartet; br, broad; m, multiplet. Mass spectra and high-resolution mass spectra were performed on a VG Micromass 7070F Mass Spectrometer with ES ionization (ESI). X-ray diffraction analysis was measured using a Bruker SMART CCD Single Crystal X-ray Diffractometer; the structure was solved by direct methods and refined on F 2 by full-matrix least-squares methods using SHELXL-97 (Sheldrick 2008). Melting points are uncorrected and expressed in °C. Optical rotations analyses were performed on a Perkin-Elmer Model 343 Polarimeter. High-resolution mass spectra were performed on a VG Micromass 7070F Mass Spectrometer with ES ionization (ESI). All commercially available reagents were used as received. Products were purified by flash column chromatography on silica gel purchased from Qingdao Haiyang Chemical Co., Ltd. All reactions involving air or moisture sensitive species were performed in oven-dried glassware under inert atmosphere.
Typical procedure for cis -2 or cis -3
To a mixture of monoacid cis -1 (1.52 g, 5.2 mmol) and l-phenylalanine ethyl ester hydrochloride (1.26 g, 5.5 mmol) in dry CH2Cl2 (30.0 mL), Et3N (2.0 mL, 14.2 mmol) was added at −5°C under nitrogen, and the mixture was stirred for 1.0 h at r.t., dicyclohexylcarbodiimide (DCC) (1.6 g, 7.5 mmol) and DMAP (60 mg, 0.5 mmol) were added to the mixture at −5°C and the mixture was stirred overnight. After the reaction was finished, it was filtered on a Celite pad. The filtrate was evaporated to give cis -2 (diastereomeric mixture) as yellow oil. The residue was purified by silica gel column chromatography to give the desired compounds (−)-4a and (−)-4b.
(−)-4a, n-hexane/ethyl acetate = 3:1 (v/v), R f = 0.45, yield: 1.02 g (41% yield), light yellow oil; (−)-4b, n-hexane/ethyl acetate = 3:1 (v/v), R f = 0.35, yield: 1.24 g (49% yield), light yellow oil.
The coupling product cis -3 (diastereomeric mixture, 2.25 g, 95% yield) was obtained by the similar procedure from d-phenylalanine ethyl ester hydrochloride and monoacid cis -1, and it could not be separated by a column chromatography.
Spectroscopic data for compound: cis -3 (diastereomeric mixture)
1H NMR (500 MHz, CDCl3): major isomer: δ 1.10–1.13 (m, 2.5H), 1.20–1.30 (m, 4H), 1.30–1.40 (m, 2.5H), 1.51–1.59 (m, 1H), 1.72–1.88 (m, 1H), 2.01 (m, 1H), 3.06–3.15 (m, 1H), 3.30–3.38 (m, 1H), 3.51–3.54 (m, 1H), 3.58–3.70 (m, 1H), 3.80–3.90 (m, 1H), 3.91–4.02 (m, 1H), 4.13–4.25 (m, 1H), 4.97 (m, 0.74H), 7.20–7.42 (m, 10H), 8.86 (m, 0.73H). Minor isomer: δ 4.65–4.75 (m, 0.20 H), 8.88 (m, 0.20 H), the other signals are overlapped with the major isomer. HRMS (ESI) m/z calcd for C27H35N2O5 (M+H): calcd 467.2546, obs 467.2540. Light yellow oil, \( [\alpha ]_{\text{D}}^{25} = - 21.4^\circ \) (c = 0.58, CHCl3).
Spectroscopic data for compound: (−)-4a
1H NMR (500 MHz, CDCl3): δ 1.24 (t, J = 7.0 Hz, 3H), 1.29 (t, J = 7.0 Hz, 3H), 1.41 (d, J = 6.5 Hz, 3H), 1.74–1.91 (m, 4H), 3.06 (dd, J 1 = 8.5 Hz, J 2 = 5.5 Hz, 1H), 3.31 (dd, J 1 = 8.5 Hz, J 2 = 5.5 Hz, 1H), 3.62 (dd, J 1 = 7.5 Hz, J 2 = 2.5 Hz, 1H), 3.68 (dd, J 1 = 5.5 Hz, J 2 = 2.5 Hz, 1H), 3.94 (q, J = 7.0 Hz, 1H), 4.08–4.17 (m, 2H), 4.25 (q, J = 7.5 Hz, 2H), 4.92–4.97 (m, 1H), 7.20–7.30 (m, 10H), 8.83 (d, J = 9.0 Hz, 1H). 13C NMR (300 MHz, CDCl3): δ 14.2, 14.3, 18.8, 29.9, 30.8, 38.3, 52.8, 60.6, 60.9, 61.2, 63.8, 64.2, 126.7, 127.4, 127.8, 128.2, 128.4, 129.2, 136.7, 141.7, 171.7, 175.1, 175.7. HRMS (ESI) m/z calcd for C27H35N2O5 (M+H): calcd 467.2546, obs 467.2542. Yellow oil, \( [\alpha ]_{\text{D}}^{25} = - 32.4^\circ \) (c = 1.06, CHCl3).
Spectroscopic data for compound: (−)-4b
1H NMR (500 MHz, CDCl3): δ 1.14 (t, J = 7.0 Hz, 6H), 1.25 (t, J = 7.0 Hz, 3H), 1.69–2.01 (m, 4H), 3.10 (dd, J 1 = 9.5 Hz, J 2 = 5.0 Hz, 1H), 3.32 (dd, J 1 = 8.5 Hz, J 2 = 5.5 Hz, 1H), 3.58 (q, J = 7.0 Hz, 2H), 3.68 (q, J = 7.0 Hz, 1H), 3.97 (m, 2H), 4.01–4.22 (m, 2H), 4.80–4.84 (m, 1H), 7.14–7.40 (m, 10H), 8.76 (d, J = 8.0 Hz, 1H). 13C NMR (300 MHz, CDCl3): δ 14.1, 14.2, 20.9, 30.6, 31.0, 37.6, 53.4, 60.8, 61.2, 62.8, 64.9, 66.4, 126.9, 127.5, 128.0, 128.3, 128.5, 129.2, 136.9, 142.4, 171.8, 175.8, 176.1. HRMS (ESI) m/z calcd for C27H35N2O5 (M+H): calcd 467.2546, obs 467.2550. Yellow oil, \( [\alpha ]_{\text{D}}^{25} = - 89.4^\circ \) (c = 1.23, CHCl3).
Typical procedure for unnatural dipeptide (+)-5a or (+)-5b
In the presence of Pd(OH)2/C (0.12 g), the compound (−)-4a (0.52 g, 1.11 mmol) in MeOH (6.0 mL) was stirred overnight under 1.0 atm H2 at r.t. After the reaction was finished, it was filtered on a Celite pad to remove catalyst. The filtrate was evaporated to give the desired product (+)-5a (yellow crystal, 0.38 g, 96% yield) without further purification.
Compound (+)-5b was obtained from (−)-4b by the similar procedure in 94% yield (white crystal, 0.43 g).
Spectroscopic data for compound: (+)-5a
1H NMR (500 MHz, CDCl3): δ 1.27 (t, J = 7.0 Hz, 3H), 1.29 (t, J = 7.0 Hz, 3H), 1.53–1.61 (m, 2H), 1.82–1.90 (m, 1H), 2.01–2.08 (m, 1H), 2.86 (br, 1H), 3.03 (dd, J 1 = 8.1 Hz, J 2 = 5.7 Hz, 1H), 3.21 (dd, J 1 = 8.1 Hz, J 2 = 6.0 Hz, 1H), 3.82–3.87 (m, 1H), 3.91–3.96 (m, 1H), 4.12–4.23 (m, 1H), 4.83–4.91 (m, 1H), 7.18–7.28 (m, 5H), 8.50 (d, J = 8.4 Hz, 1H). 13C NMR (300 MHz, CDCl3): δ 14.1, 14.3, 29.6, 31.2, 38.2, 52.9, 60.3, 61.0, 61.2, 61.3, 126.7, 128.2, 128.3, 129.3, 136.5, 171.8, 174.5, 174.9. HRMS (ESI) m/z calcd for C19H27N2O5 (M+H): calcd 363.1920, obs 363.1929. Yellow crystal, m.p. 54.5–56.0°C, \( [\alpha ]_{\text{D}}^{25} = + 4.5^\circ \) (c = 1.0, CHCl3).
Spectroscopic data for compound: (+)-5b
1H NMR (300 MHz, CDCl3): δ 1.22 (t, J = 7.2 Hz, 3H), 1.27 (t, J = 7.2 Hz, 3H), 1.90–2.17 (m, 4H), 2.66 (br, 1H), 3.06 (dd, J 1 = 8.7 Hz, J 2 = 5.7 Hz, 1H), 3.20 (dd, J 1 = 8.4 Hz, J 2 = 5.4 Hz, 1H), 3.75–3.85 (m, 1H), 3.90–3.96 (m, 1H), 4.12–4.23 (m, 1H), 4.68–4.75 (m, 1H), 7.21–7.32 (m, 5H), 8.55 (d, J = 7.5 Hz, 1H). 13C NMR (300 MHz, CDCl3): δ 14.1, 14.2, 29.8, 31.4, 37.8, 53.6, 60.3, 61.1, 61.4, 126.8, 128.4, 129.0, 129.3, 136.8, 171.5, 174.6, 175.5. HRMS (ESI) m/z calcd for C19H27N2O5 (M+H): calcd 363.1920, obs 363.1893. White crystal, m.p. 81.0–82.0°C, \( [\alpha ]_{\text{D}}^{25} = + 13.9^\circ \) (c = 1.05, CHCl3).
Typical procedure for unnatural dipeptide (−)-6a or (−)-6b
The compound (−)-4a (0.95 g, 2.1 mmol) in the mixed solvent of THF and H2O (10.0 mL, v/v = 1:1) was added by KOH pellets (0.36 g, 5.1 mmol) and the mixture was stirred 2 h at r.t. After the reaction was finished, the solvent was evaporated and the acidity of the aqueous residue was adjusted to be pH 3.0 by 6.0 M HCl, then it was extracted by ethyl acetate (3× 10 mL), the combined organic layer was washed by H2O (2× 5 mL) and brine (10 mL), dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure to give the desired product (−)-6a as white powder (0.83 g, 96% yield) without further purification.
Compound (−)-6b was obtained from (−)-4b by the similar procedure in 94% yield (white crystal, 0.80 g).
Spectroscopic data for compound: (−)-6a
1H NMR (500 MHz, DMSO-D6): δ 1.27 (d, J = 7.0 Hz, 3H), 1.30 (m, overlap, 1H), 1.45 (m, 1H), 1.77–1.89 (m, 2H), 2.83 (dd, J 1 = 10.0 Hz, J 2 = 4.0 Hz, 1H), 3.17 (dd, J 1 = 9.5 Hz, J 2 = 4.5 Hz, 1H), 3.47 (m, 1H), 3.59 (m, 1H), 3.90 (m, 1H), 4.57–4.61 (m, 1H), 7.18–7.32 (m, 10H), 8.79 (d, J = 9.0 Hz, 1H), 12.8 (br, 2H). 13C NMR (300 MHz, CDCl3): δ 20.9, 29.9, 30.6, 37.9, 53.1, 61.4, 64.5, 64.7, 126.9, 127.5, 127.9, 128.5, 128.7, 129.4, 143.5, 173.3, 174.6, 177.5. HRMS (ESI) m/z calcd for C23H27N2O5 (M+H): calcd 411.1920, obs 411.1912. White powder, m.p. 193.5–194.5°C, \( [\alpha ]_{\text{D}}^{25} = - 43.4^\circ \) (c = 0.55, DMSO).
Spectroscopic data for compound: (−)-6b
1H NMR (500 MHz, DMSO-D6): δ 1.03 (d, J = 7.0 Hz, 3H), 1.69–1.80 (m, 2H), 1.89–1.99 (m, 2H), 2.85 (dd, J 1 = 11.0 Hz, J 2 = 3.0 Hz, 1H), 3.22 (dd, J 1 = 9.5 Hz, J 2 = 4.5 Hz, 1H), 3.44 (m, 1H), 3.51 (m, 1H), 3.72 (m, 1H), 4.39–4.44 (m, 1H), 7.14–7.36 (m, 10H), 8.80 (d, J = 7.5 Hz, 1H), 12.6 (br, 2H). 13C NMR (300 MHz): δ 20.8, 29.6, 31.0, 38.2, 52.8, 60.6, 63.9, 64.2, 126.7, 127.2, 128.0, 128.7, 128.9, 129.2, 142.9, 172.3, 174.8, 178.0. HRMS (ESI) m/z calcd for C23H27N2O5 (M+H): calcd 411.1920, obs 411.1914. White powder, m.p. 201.0–203.0°C, \( [\alpha ]_{\text{D}}^{25} = - 60.8^\circ \) (c = 0.4, DMSO).
Typical procedure for unnatural dipeptide (+)-7a or (+)-7b
In the presence of Pd(OH)2/C (0.13 g), the compound (−)-6a (0.61 g, 1.5 mmol) in MeOH (6.0 mL) was stirred overnight under 1.0 atm H2 at r.t. After the reaction was finished, it was filtered on a Celite pad to remove catalyst. The filtrate was evaporated to give the desired product (+)-7a as white powder (0.42 g, 91% yield) without further purification.
Compound (+)-7b was obtained from (−)-6b by the similar procedure in 89% yield (white powder, 0.40 g).
Spectroscopic data for compound: (+)-7a
1H NMR (300 MHz, DMSO-D6): δ 1.49–1.61 (m, 1H), 1.67–1.80 (m, 1H), 1.95–2.11 (m, 2H), 2.63 (dd, J 1 = 8.7 Hz, J 2 = 5.1 Hz, 1H), 2.83 (dd, J 1 = 7.8 Hz, J 2 = 5.7 Hz, 1H), 3.71–3.75 (m, 1H), 3.80–3.85 (m, 1H), 3.88–3.96 (m, 1H), 4.94 (br, 1H), 7.15–7.28 (m, 5H), 8.38 (d, J = 8.4 Hz, 1H). 13C NMR (300 MHz, CDCl3): δ 30.2, 31.4, 38.8, 54.3, 61.8, 62.4, 127.1, 128.6, 129.2, 129.6, 141.5, 172.3, 176.6, 177.2. HRMS (ESI) m/z calcd for C15H19N2O5 (M+H): calcd 307.1294, obs 307.1298. White powder (very hygroscopic), \( [\alpha ]_{\text{D}}^{25} = + 11.6^\circ \) (c = 0.5, DMSO).
Spectroscopic data for compound: (+)-7b
1H NMR (300 MHz, DMSO-D6): δ 1.25–1.30 (m, 1H), 1.70–1.77 (m, 1H), 1.97–2.05 (m, 2H), 2.55–2.63 (m, 1H), 2.85–2.91 (m, 1H), 3.67–3.71 (m, 1H), 3.88–4.02 (m, 2H), 4.96 (br, 1H), 7.14–7.27 (m, 5H), 8.42 (d, J = 8.7 Hz, 1H). 13C NMR (300 MHz, CDCl3): δ 28.2, 32.1, 37.4, 55.6, 60.9, 61.7, 126.9, 127.8, 128.8, 129.5, 139.8, 171.4, 175.7, 176.1. HRMS (ESI) m/z calcd for C15H19N2O5 (M+H): calcd 307.1294, obs 307.1290. White powder (very hygroscopic), \( [\alpha ]_{\text{D}}^{25} = + 25.2^\circ \) (c = 0.5, DMSO).
Result and discussion
The monoacid cis -1 was obtained in 76% yield by previously reported protocol of selective monohydrolysis (Wang et al. 2007) of diethyl cis-1-[(S)-1-phenylethyl]pyrrolidine-2,5-dicarboxylate (Yamamoto et al. 1993). The coupling reactions of monoacid cis -1 with l- and d-phenylalanine ethyl ester hydrochloride were investigated, respectively. In the presence of triethylamine, the coupling of phenylalanine ethyl ester hydrochlorides with monoacid cis -1 was performed smoothly using 1.5 equiv. DCC as coupling reagent and CH2Cl2 as solvent at room temperature (Scheme 1). Both cis -2 and cis -3 were obtained in excellent yields (up to 95%). To our delight, the diastereomeric mixture cis -2 prepared from l-phenylalanine ethyl ester and monoacid cis -1 was easily separated to be (−)-4a and (−)-4b by a flash column chromatography (FC) on silica gel; however, the diastereomeric mixture cis -3 prepared from monoacid cis -1 and d-phenylalanine ethyl ester instead could not be separated by the column chromatography (Scheme 2). We could deduce from 1H NMR spectrum that the diastereomeric ratio of the major and the minor in cis -3 is about 3.5/1.

Synthesis of cis -2 and cis -3

Synthetic routes to unnatural dipeptides (+)-5a and (+)-5b
Consequently, compounds (−)-4a and (−)-4b were hydrogenolysed using catalytic amount of Pd(OH)2/C in methanol to afford the corresponding dipeptides (+)-5a and (+)-5b with protected carboxylic groups, respectively (Scheme 2). The dipeptides (+)-5a and (+)-5b containing cis-pyrrolidine structure with free N-terminal at pyrrolidine ring can be served as valuable building blocks for connection of other amino acids to furnish some other complicated peptides.
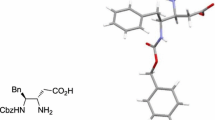
Fortunately, compound (+)-5a got crystallized from CH2Cl2. The X-ray diffraction analysis (Fig. 2) shows the cis-configuration at C4 and C7. The crystal packing is found to be orthorhombic. The absolute configurations of C4, C7 and C9 of (+)-5a were deduced from the starting material l-phenylalanine, which should be S, R and S, separately. Some crystal data of (+)-5a are listed in Table 1.

ORTEP diagram of compound (+)-5a
Hydrolysis of compounds (−)-4a and (−)-4b using solid KOH in THF-H2O afforded the corresponding dipeptides (−)-6a and (−)-6b with two free C-termini (Scheme 3) and these free carboxylic groups provide the possibilities for bonding some other amino acids to yield complex unnatural peptides. The other two unnatural dipeptides (+)-7a and (+)-7b with both free C- and N-terminus were obtained by Pd(OH)2/C catalytic hydrogenolysis of compounds (−)-6a and (−)-6b in methanol at room temperature separately. The compounds (+)-7a and (+)-7b found to be hygroscopic.

Synthetic routes to unnatural dipeptides (+)-7a and (+)-7b
Conclusion
In summary, we provide a simple and efficient construction of unnatural dipeptides based on cis-2,5-disubstituted pyrrolidine backbone from commercially available starting materials, and these unnatural peptides contain free C-terminus or N-terminus which are convenient to couple with the other amino acids to give more complex polypeptides. The configurations of these unnatural dipeptides are confirmed by X-ray crystal diffraction analysis. The synthesis of some novel bioactive cyclic peptides derived from these unnatural dipeptides is currently investigated in our laboratory.
References
Bagley MC, Tovey J (2001) Diastereoselective synthesis of cis-2,5-disubstituted pyrrolidine N-oxides by the retro-Cope elimination. Tetrahedron Lett 42:351–353
Brenneman JB, Martin SF (2004) Application of intramolecular enyne metathesis to the synthesis of aza[4.2.1]bicyclics: enantiospecific total synthesis of (+)-Anatoxin-a. Org Lett 6:1329–1331
Cahill JP, Cunneen D, Guiry PJ (1999) trans-2,5-Dialkylpyrrolidinyl-containing phosphinamines. Synthetic and mechanistic studies in Pd-catalysed asymmetric allylic alkylation. Tetrahedron Asymmetry 10:4157–4173
Chen HF, Sweet JA, Lam KC, Rheingold AL, McGrath DV (2009) Chiral amine–imine ligands based on trans-2,5-disubstituted pyrrolidines and their application in the palladium-catalyzed allylic alkylation. Tetrahedron Asymmetry 20:1672–1682
Colandrea VJ, Legiec IE, Huo P, Yan L et al (2006) 2,5-Disubstituted pyrrolidine carboxylates as potent, orally active sphingosine-1-phosphate (S1P) receptor agonists. Bioorg Med Chem Lett 16:2905–2908
Davis FA, Zhang JY, Qiu H, Wu YZ (2008) Asymmetric synthesis of cis- and trans-2,5-disubstituted pyrrolidines from 3-oxo pyrrolidine 2-phosphonates: synthesis of (+)-Preussin and analogs. Org Lett 10:1433–1436
Dhimane H, Vanucci C, Lhommet G (1997) Stereoselective access to trans-2,5-disubstituted pyrrolidine derivatives by nucleophilic addition to bicyclic N-acyliminium ion. Tetrahedron Lett 38:1415–1418
Draper JA, Britton R (2010) A concise and stereoselective synthesis of hydroxypyrrolidines: rapid synthesis of (+)-Preussin. Org Lett 12:4034–4037
Enache LA, Kennedy I, Sullins DW, Chen W, Ristic D et al (2009) Development of a scalable synthetic process for DG-051B, A first-in-class inhibitor of LTA4H. Org Process Res Dev 13:1177–1184
Giordano C, Sansone A, Masi A, Lucente G et al (2010) Synthesis and activity of endomorphin-2 and morphiceptin analogues with proline surrogates in position 2. Eur J Med Chem 45:4594–4600
Haddad M, Hassan Imogaï H, Larchevêque M (1998) The first enantioselective synthesis of the cis-2-carboxy-5-phenylpyrrolidine. J Org Chem 63:5680–5683
Hanessian S, Auzzas L (2008) The practice of ring constraint in peptidomimetics using bicyclic and polycyclic amino acids. Acc Chem Res 41:1241–1251
Isidro-Llobet A, Murillo T, Bello P, Cilibrizzi A, Hodgkinson JT et al (2011) Diversity-oriented synthesis of macrocyclic peptidomimetics. Proc Natl Am Sci. doi:10.1073/pnas.1015267108
Jefford CW, Tang Q, Zaslona A (1991) Short, enantiogenic syntheses of (−)-indolizidine 167B and (+)-monomorine. J Am Chem Soc 113:3513–3518
Lee E, Jeong EJ, Min SJ, Hong SK et al (2000) Radical cyclization of β-aminoacrylates: synthesis of (−)-indolizidine 223AB. Org Lett 2:2169–2171
Lemen GS, Wolfe JP (2010) Pd-catalyzed carboamination of oxazolidin-2-ones: a stereoselective route to trans-2,5-disubstituted pyrrolidines. Org Lett 12:2322–2325
Lewin AH, Naseree T, Ivy Carroll F (1987) A practical synthesis of (+)-cocaine. J Heterocyclic Chem 24:19–21
Lygo B, Beynon C, Michael C, McLeod MC, Roy CE, Wade CE (2010) Application of asymmetric phase-transfer catalysis in the enantioselective synthesis of cis-5-substituted proline esters. Tetrahedron 66:8832–8836
Madis V (1977) Flexibility of the pyrrolidine ring in proline peptides. Biopolymers 16:2671–2692
Manfré F, Pulicani JP (1994) Enantiospecific synthesis and absolute configuration of (+)-RP 66803 a new non-peptide CCK antagonist. Tetrahedron Asymmetry 5:235–238
Mans DM, Pearson WH (2004) Total synthesis of (+)-cocaine via desymmetrization of a meso-dialdehyde. Org Lett 6:3305–3308
Miao L, Shu H, April R, Noble AR, Steven P, Fournet SP et al (2010) An enantioselective formal synthesis of (+)-Gephyrotoxin 287C. ARKIVOC iv:6–14
Pei ZH, Li XF, Longenecker K, von Geldern TW et al (2006) Discovery, structure–activity relationship, and pharmacological evaluation of (5-substituted-pyrrolidinyl-2-carbonyl)-2-cyanopyrrolidines as potent dipeptidyl peptidase IV inhibitors. J Med Chem 49:3520–3535
Sheldrick GM (2008) A short history of SHELX. Acta Cryst A 64:112–122
Shu H, Noble AR, Zhang SH, Miao L, Trudel ML (2010) Enantioselective syntheses of both enantiomers of cis-pyrrolidine 225H. Tetrahedron 66:4428–4433
Sun HY, Nikolovska-Coleska Z, Yang CY, Qian DG, Lu JF et al (2008) Design of small-molecule peptidic and nonpeptidic smac mimetics. Acc Chem Res 41:1264–1277
Takahata H, Hiroyuki Takehara H, Ohkubo N, Momose T (1990) An efficient route to chiral trans-2,5-dialkylpyrrolidines via stereoselective intramolecular amidomercuration. Tetrahedron Asymmetry 1:561–566
Ung P, Winkler DA (2011) Tripeptide motifs in biology: targets for peptidomimetic design. J Med Chem 54:1111–1125
Vaswani RG, Limon A, Reyes-Ruiz JM, Miledi R, Chamberlin AR (2009) Design, synthesis, and biological evaluation of a scaffold for iGluR ligands based on the structure of (−)-kaitocephalin. Bioorg Med Chem Lett 19:132–135
Wang Q, Sasaki NA, Riche C, Potier P (1999) A versatile method for the facile synthesis of enantiopure trans- and cis-2,5-disubstituted pyrrolidines. J Org Chem 64:8602–8607
Wang Q, Tran Huu Dau M-E TH, Sasaki NA, Potier P (2001) Facile synthesis of N-Boc-(2S,5R)-5-(1′-hydroxy-1′-methylethyl)proline. Tetrahedron 57:6455–6462
Wang PA, Xu ZS, Chen CF, Gao XG, Sun XL, Zhang SY (2007) Facile synthetic route to enantiopure unsymmetric cis-2,5-disubstituted pyrrolidines. Chirality 19:581–588
Warren JG, Revilla-Lpez G, Alemn C, Jimnez AI, Cativiela C, Torras J (2010) Conformational preferences of proline analogues with a fused benzene ring. J Phys Chem B 114:11761–11770
Yamamoto Y, Hoshino J, Fujimoto Y, Ohmoto J, Sawada S (1993) A convenient synthesis of enantiomeric pairs of 2,5-disubstituted pyrrolidines of C2-symmetry. Synthesis 3:298–302
Acknowledgments
We would like to thank the National Science Foundation of China (No. 20802092) for financial support of this work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Wang, PA., He, W., Cheng, SK. et al. Efficient synthesis of unnatural dipeptides based on cis-2,5-disubstituted pyrrolidine. Amino Acids 42, 2121–2127 (2012). https://doi.org/10.1007/s00726-011-0949-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-011-0949-4