
Abstract
Manganese(III) (3d4, S = 2) coordination complexes have been widely studied by high-frequency and -field EPR (HFEPR) for their own inherent chemical interest and for providing information for the burgeoning area of molecular magnetism. In the present study, we demonstrate how a stable, easily handled complex of MnIII, [MnLKNO3], where L3− is a hexadentate tripodal ligand, trianion of 1,1,1-tris[(3- methoxysalicylideneamino)methyl]ethane, can be used for another purpose entirely. This purpose is as a field and frequency standard for HFEPR that is superior to a “traditional” standard such as an organic radical (e.g., DPPH) with its single, g = 2.00 signal, or to atomic hydrogen, which is less readily available than DPPH and provides only two signals for calibration purposes (Stoll et al. in J Magn Reson 207:158–163, 2010). By contrast, polycrystalline [MnLKNO3] (1) orients in the external magnetic field of an HFEPR spectrometer (three different spectrometers were employed in this study). The crystal structure of 1 allows determination of the exact, reproducible molecular orientation of 1 in the applied field. This phenomenon provides multiple, well-defined resonances over a broad field sweep range (0–36 T) at any of a wide range of frequencies (tested up to 1 THz so far) allowing accurate calibration of magnetic field in a multi-frequency HFEPR study.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Electron Paramagnetic Resonance (EPR) is an acknowledged magnetic resonance technique to investigate transition metal complexes [1]. In case of high-spin (S > 1/2) metal ions, high-frequency and -field EPR (HFEPR) is often required because of the phenomenon of zero-field splitting (zfs) that renders many paramagnetic complexes “EPR-silent” at conventional frequencies (X-, Q- or even W-band). A side effect of performing an HFEPR experiment on solid (polycrystalline) coordination complexes is torquing or aligning of the crystallites in the magnetic field. Any magnetic dipole inserted in a static magnetic field will undergo a torquing effect, as has been known to sailors using compasses since antiquity. In the case of paramagnets, however, the magnetic moment is much smaller than in ferromagnets, so the effect does not show up prominently. It was observed long ago [2] that crystals of paramagnetic metal coordination complexes behave as weak magnetic dipoles and, as such, they may undergo magnetic torquing if the applied field is strong enough or the temperature low enough. Also important is magnetic anisotropy. In the absence of magnetic anisotropy, the moment will align with the field. Consequently, there will be no torque on the sample/crystal. It is in the presence of magnetic anisotropy that the torque on the sample/crystal emerges. The main point is that, when the temperature is lower than the magnetic anisotropy energy scale, the magnetic moment remains locked into a preferred orientation that is related to the molecular/crystal structure. If this direction is not aligned with the applied field, there is then a torque on the sample/crystal. It is this effect that causes the alignment. When working with polycrystalline materials, the effect can be either beneficial or detrimental for an HFEPR experimentalist. In the best-case scenario, the crystallites orient such that their principal anisotropy axis is parallel to the applied field, which can be identified and attributed to the molecular structure. In a less-than-optimal situation, the crystallites orient incompletely, yielding uninterpretable spectra, or orient along a direction that cannot be attributed to the molecular structure, e.g., due to crystal shape anisotropy.
In polycrystalline transition metal complexes, the torquing cannot be dependably predicted as it depends on multiple factors, both microscopic, such as the zero-field splitting tensor of spin S > 1/2 systems (or g-anisotropy in the case of S = 1/2 systems), and macroscopic, such as the size and shapes of the crystallites, mechanical and electrostatic interactions among them, resulting in an overall macroscopic magnetic moment of a crystallite. Concentrating on coordination complexes of the high-spin (S = 2) manganese(III) ion, the “deliciae of an EPR spectroscopist” [3], some of them measured by HFEPR as solids undergo torquing, and some do not. Thus, in one of the two earliest HFEPR reports on high-spin MnIII, some of us presented field-aligned spectra of five-coordinate porphyrinato complexes [4]. The other 1997 paper by Barra et al. [5], on the other hand, presented and interpreted what were apparently powder-patterned spectra of the complex Mn(dbm) (dbm = anion of 1,3-diphenyl-1,3-propanedione (dibenzoylmethane)). Similar to the latter, the complex [Mn(dbm)2(py)2](ClO4) (py = pyridine; trans-N2O4 donor set) reported by some of us [6] offered a case of almost ideal HFEPR powder patterns without constraining. Note that field-induced torquing is not restricted to mononuclear complexes; some of us (and many others) reported on polynuclear complexes of manganese exhibiting the same effect [7].
In this work, we discuss a high-spin MnIII complex denoted as [MnLKNO3] where L3− is a hexadentate tripodal ligand, the trianion of 1,1,1-tris[(3- methoxysalicylideneamino)methyl]ethane, which presents a facial N3O3 donor set to the MnIII ion. A molecular structure drawing is shown in Scheme 1.

Structure of [MnLKNO3] (1) with the K+ and NO3- ions omitted for clarity (see Fig. 1 for complete structure from X-ray crystallography)
Despite having very similar microscopic parameters to both the aforementioned dbm-ligated complexes (i.e., negative D in the 4.5–5 cm–1 range and small |E|, g ~ 1.98), [MnLKNO3] (1) undergoes complete torquing. We will show and discuss below how this phenomenon can be used to the advantage of ultra-high HFEPR performed at frequencies reaching 1 THz and fields up to 36 T. We note at this point that the frequency of 1 THz is an “overkill” as an S = 2 spin system characterized by the zfs of this order of magnitude will show multiple transitions down to about ν ~ D/h, i.e., ~ 100 GHz. It will most likely show a single resonance at X- or Q-band as well, corresponding to the nominally forbidden ΔMS = 4 transition [8].
2 Experimental Section
2.1 Synthesis
Complex 1 was prepared by the 1:1:1 reaction of H3L, MnII(OAc)2 ‧ 4H2O and KNO3 in methanol in air and obtained as deep green needles. Elemental analysis results and IR spectra revealed that the product was [MnLKNO3] [9].
2.2 X-Ray Diffraction
A green block crystal of 1 having approximate dimensions of 0.220 × 0.130 × 0.100 mm3 was mounted on a glass fiber. All measurements were made on a Rigaku R-AXIS RAPID diffractometer at 180 K using graphite-monochromated Mo-Kα radiation. Of the 28,109 reflections collected, 6702 were unique (Rint = 0.0412); equivalent reflections were merged. The linear absorption coefficient, μ, for Mo-Kα radiation is 6.556 cm–1. A numerical absorption correction was applied which resulted in transmission factors ranging from 0.883 to 0.937. The data were corrected for Lorentz and polarization effects. The structure was solved by direct methods [10] and expanded using Fourier techniques. The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were refined using the riding model. The final cycle of full-matrix least-squares refinement [11, 12] on F2 was based on 6702 observed reflections and 397 variable parameters. All calculations were performed using the CrystalStructure [13] crystallographic software package except for refinement, which was performed using SHELX97 [11, 12]. The originally reported structure is CCDC 812377 (CSD code IXUSUQ) [9]. This structure was revised by removing several bad reflections according to recent checkCIF results to give the structure reported herein (CCDC 2355722).
2.3 HFEPR
Three different HFEPR spectrometers/magnets were used in this work. The one within the NHMFL EMR Facility is based on a 15/17 T superconducting magnet and has been described previously [14], but now with an addition of more recent solid-state source chains from VDI (Virginia Diodes Inc., Charlottesville, VA, USA) operating up to ~ 640 GHz. Two other setups are part of the NHMFL DC Facility: one was based on a 25 T resistive magnet with improved homogeneity (the so called “Keck” magnet; now decommissioned) and Backward Wave Oscillators (BWOs) employed as sources operating up to 1 THz, as described by Zvyagin et al. [15]. The most recent addition to the NHMFL magnetic resonance armamentarium is a spectrometer based on the Series Connected Hybrid (SCH) 36 T magnet, employing a different VDI chain operating up to 1 THz, as described by Dubroca et al. [16].
The spectra were interpreted via a standard spin Hamiltonian for S = 2:
where \(\beta\) is the Bohr magneton, \(\widehat{S}\) is the vector spin operator (with components \({\widehat{S}}_{i}, i=x, y, z\)), \({\varvec{g}}\) is the electron \({\varvec{g}}\)-tensor, \(\mathop{H}\limits^{\rightharpoonup}\) is the applied external magnetic field, and D and E are axial and rhombic zero-field splitting (zfs) parameters, respectively.
The simulations and fitting were achieved using the SPIN package written by A. Ozarowski [17].
3 Results and Discussion
3.1 Crystal Structure
The single-crystal X-ray diffraction (XRD) structure of 1 was reported previously [9], but is now revised by removing several bad reflections according to recent checkcif results. There are no significant structural differences between the previously reported and revised structures. The molecule crystallizes in a monoclinic space group P21/c with four molecules in the unit cell. The structure of 1 is reproduced below in Fig. 1. Axial elongation is observed along the O3–Mn1–N2 direction, as a consequence of the Jahn–Teller effect as is typically seen in high-spin d4 complexes, such as of MnIII, and was extensively discussed previously [9]. A potassium cation is in the O6 pocket of the [MnL] moiety with an average K–O distance of 2.8112 Å. A nitrate is connected to K+ with an average K–O distance of 2.7784 Å. Due to the presence of K+, the Mn–O and Mn–N distances of 1 are longer by 0.0032 Å on average than those of K+-free [MnL] which also contains an axially elongated MnIII [9].

X-ray diffraction-obtained molecular structure of 1. The relevant metrics (in Å) are: Mn(1)—O(1): 1.8969(13); Mn(1)—O(2): 1.9383(13); Mn(1)—O(3): 2.0612(13); Mn(1)—N(1):2.0195(16); Mn(1)—N(2): 2.2177(17); Mn(1)—N(3): 2.0583(16). The especially elongated Mn1–N2 bond distance is indicated in boldface
3.2 High-Frequency and -Field EPR
An HFEPR spectrum of complex 1 “as is”, i.e., as obtained from the last crystallization, recorded at 638 GHz and 4.2 K is shown in Fig. 2 (top, black). The spectrum suffers from what is sometimes colloquially called a “grass pattern”, i.e., showing individual crystallites instead of their powder distribution, and is uninterpretable. Reducing the size of crystallites by repeated grinding resulted in a spectrum shown in Fig. 2 (middle, blue) showing well-defined resonances. The derivative shape of these resonances suggests that the spectrum does not correspond to a powder pattern but is similar to what is expected from a single crystal, indicating a presence of strong torquing in the magnetic field. The spectrum by itself is not informative as to the spin Hamiltonian parameters, as proved by a futile attempt to simulate the frequency dependence of resonances as shown in Fig. 3 (assuming these resonances correspond to canonical orientations of the zfs with regard to the field). This failure contrasts with many successful examples wherein least-squares fitting of experimental points leads to a perfect match between experimental features and calculated curves. The plots shown in Fig. 3 indicate, however, several near-zero-field resonances at ca. 95, 395, 500 and 600 GHz. Assuming those resonances represent the |D – 3E|, |3D – 3E|, |3D + 3E| and 4|D| zero-field transitions in an S = 2 spin system, “back-of-the-envelope” calculations deliver estimates of the zfs parameters: |D|= 148 GHz = 4.93 cm–1 and |E|= 17 GHz = 0.57 cm–1. These values were used to simulate the field vs. frequency dependences in Fig. 3, which do not coincide with the observation.

HFEPR spectra of complex 1 recorded at 4.2 (black trace) or 10 K (color traces) and 638 GHz using the 15 T superconducting magnet. Top black trace: sample “as is”; blue trace: sample ground; red trace: sample repeatedly ground and pressed in a pellet with n-eicosane. The amount of sample (~ 60 mg) remained the same in each experiment as were other conditions, such as receiver gain and modulation amplitude

A two-dimensional (field vs. frequency or energy) map of resonances in the loose powder spectra of 1 (squares) obtained in the resistive “Keck” magnet and its simulation (curves) using the spin Hamiltonian parameters: |D|= 4.93 cm–1, |E|= 0.57 cm–1 as estimated from the near-zero-field transitions indicated by the red arrows. The g-value was assumed as 2.00. Red curves: simulated turning points with magnetic field B0 parallel to the x-axis of the zfs tensor, blue: B0 || y, black: B0 || z. Although the simulation agrees with the observed near-zero-field resonances, it does not reproduce the positions of high-field peaks because these resonances do not correspond to the canonical orientations of the zfs tensor relative to the field expected in a powder
The final proof of the presence of torquing is obtained by pressing the powder into a pellet with n-eicosane. The resulting spectrum shows peaks of much lower amplitude (Fig. 2 bottom, red trace) due to a dispersion of resonances belonging to individual crystallites over a wide field range, which, however, can be simulated as a powder pattern at lower frequencies, where more power is available, such as 295.2 GHz (Fig. 4). Such single-frequency simulations are useful in determining the sign of D, which in this case is negative, as expected from the elongation-type Jahn–Teller distortion [18, 19] of the octahedral geometry of 1 revealed by the structural studies (Fig. 1).

A representative HFEPR spectrum of 1 as a pellet recorded at 295.2 GHz and 15 K (black trace) using the 15 T superconducting magnet and its simulations using the following spin Hamiltonian parameters: |D|= 4.84, |E|= 0.61 cm–1, giso = 1.98. Red trace: D > 0, blue trace: D < 0. The intensity of particular turning points agrees much better with the simulation for D < 0
Spin Hamiltonian parameters applied to optimally simulate single-frequency spectra are usually somewhat frequency-dependent. To establish frequency-independent parameters, the frequency dependence of the turning points in powder spectra as obtained from a pellet was compiled as a two-dimensional field vs. frequency or energy map (Fig. 5), similarly to Fig. 3, and then used for a simultaneous fit of spin Hamiltonian parameters according to the tunable-frequency methodology [20]. The parameters thus obtained are: |D|= 4.780(6), |E|= 0.590(5) cm–1, g = [2.003(5), 1.992(5), 1.970(13)], which are typical values for quasi-octahedral complexes of MnIII [6, 21].

A two-dimensional (field vs. frequency or energy) map of turning points in the pellet spectra of 1 (squares) obtained in the 15 T superconducting magnet and its simulation (curves) using the spin Hamiltonian parameters given in Table 1. Red curves: turning points with magnetic field B0 parallel to the x-axis of the zfs tensor, blue: B0 || y, black: B0 || z, green: off-axis turning points. For an explanation of the off-axis turning points [22], see the Supplementary Information
With the spin Hamiltonian values fixed from the experiments on the pellet, we returned to the spectra of the loose ground powder of 1 and found out by trial and error that they can be very satisfactorily simulated assuming that they originate from a single crystal oriented in the magnetic field in such a way that the polar angles of the zfs tensor relative to the magnetic field are: θ = 38 ± 2°, φ = 27 ± 5°. The positions of the simulated resonances depend very strongly on the angle θ but relatively weakly on φ.
The question that then needs to be answered is how these angles, particularly θ, relate to the crystal structure. The unit cell of complex 1 consists of four molecules (Z = 4) of which two are related to the other two by inversion and have the same orientation versus the magnetic field at an arbitrary crystal orientation. However, the molecules which are related by the C2 axis are oriented differently versus the field at an arbitrary crystal orientation. This means that, for an arbitrary orientation of the crystal relative to the magnetic field, there should be two sets of EPR resonances, each corresponding to one of the two differently oriented molecules, yet the experiment clearly shows a single set of resonances. We thus looked for orientations in which all the molecules would be equivalent in the magnetic field. The C2-related molecules are oriented versus the magnetic field in the same way when the magnetic field is either in the crystallographic ac plane or is parallel to the b axis (which is the C2 rotation axis) (Fig. 6). In the ac plane, the range of the angle between the magnetic field and the Mn-N2 axis, i.e., the Jahn–Teller elongation axis, which most likely corresponds to the z-axis of the zfs tensor, is 54.1° to 90° while along the b axis it is 35.9°. With a negative D, the orientation with a smaller angle is energetically favored. The experimental spectrum indeed appears to correspond to a molecular orientation with θ = 36°; thus, we conclude that the crystallites have been largely aligned with their b axes parallel to the magnetic field. This orientation corresponds to a minimum energy of two C2-related magnetic dipoles (see the Supporting Information).

A non-perspective (parallel projection) view of the Mn coordination sphere viewed along the crystallographic b axis. Magenta: Mn; red: oxygen; blue: nitrogen N1 and N3; green: nitrogen N2. The z-axis of the zfs tensor is expected to coincide or be close to the Mn-N2 direction. The original molecule specified in the CIF file is in the bottom right corner, the other one was generated by the C2 symmetry operation (2–x, ½ + y, ½–z). The magnetic field ‘sees’ both molecules in the same way. The angle between the magnetic field and the Mn-N2 vector is 35.9° in each case and only one spectrum will be seen in a single crystal oriented with its b axis parallel to the field
At this point, we would like to mention that field-induced orientation along an average direction (as opposed to a direction corresponding to the maximum anisotropy of a particular molecule) appears to be a common phenomenon when there are multiple orientations of the molecules within the unit cell as seen e.g., in a ReIV complex [23] and in a EuII complex [24]. Based on available data, it is most likely that the average direction will coincide with one of the crystal symmetry axes, in this case the b axis. What makes complex 1 unique is that the torquing effect is nearly complete, and very consistent. Various batches of the same material always yield the same spectra. The material is also very stable and has not deteriorated in 15 years since its synthesis and delivery, despite being stored in the humid Florida climate with no particular precautions.
Because of its properties, we have come to use 1 as a standard in two different situations:
-
a)
Complex 1 as a frequency standard. The BWO sources lack a frequency lock, thus their output frequency is determined by pre-calibration using a quasi-optical Fabry–Perot resonator. The accuracy of such calibration is estimated as ± 0.5% at best. This means that, in the high-frequency BWOs operating near 1 THz, the frequency is typically determined within 10 GHz. Moreover, some high-frequency BWOs have a tendency of jumping modes, deviating sometimes by several GHz from their original calibration curve. Such a jump is normally not detected directly during the experiment. A sample of 1 can then be used to re-calibrate the frequency. As opposed to other standards typically used in EPR, such as DPPH or BDPA, which produce only a single line near g = 2.00, or the atomic hydrogen standard, available in a stable form, which produces only two lines with exactly known splitting (1416.8 MHz, when below 70 K) centered at the g = 2.00 field position [25], complex 1 generates a series of resonances spread over a wide range of magnetic field. This behavior is particularly useful when operating at high frequencies in a superconducting magnet where the field is relatively well known but has a limit of typically 15–16 T (see below). Since 16 T limits the frequency of a g = 2.00 standard to ~ 447 GHz, working at a higher frequency will not benefit from the g = 2.00 standards. We have previously published a spectrum of 1 recorded at 936 GHz (see Fig. 4 of Krzystek and Telser [26]). In that case, prior knowledge of the spin Hamiltonian parameters and the orientation of the sample allowed us to correct the BWO frequency by 9 GHz (~ 1%), the deviation caused by mode jumping in that particular tube.
-
b)
Complex 2 as a field standard. While superconducting magnets can be calibrated within reasonable accuracy, this is not the case with resistive magnets such as the ones used at the NHMFL to perform ultra-high-field EPR. These are typically calibrated using NMR at a few discrete points, and only at their commissioning or after a major repair and rebuild. This problem is also present in the newest addition to the EPR operation at the NHMFL, the 36 T series-connected hybrid (SCH) magnet-based spectrometer [16]. Figure 7 top (upper, black trace) shows a spectrum of 1 recorded in that magnet using a VDI chain locked at 945.360(7) GHz. The middle (blue) trace is its single-crystal simulation at the orientation given by the polar coordinates (θ = 36, φ = 27 deg). The θ value is taken from the crystal structure as the angle between the b axis and the Jahn–Teller elongation direction, the φ value is determined by trial and error, as described before. The bottom red trace represents the same experimental file as the top black one but after adjusting the magnetic field according to the formula Badj = Bexp – i × 8.41 μT, where i is the point number, 0 < i < 60,000, resulting in a much better agreement with the simulation. This result suggests that broad-sweep spectra taken in the SCH magnet should have their field scale adjusted by reducing the field correspondingly. The above formula was determined empirically and is valid only for the specific magnet, sweep range and rate.

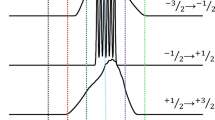
Upper panel: spectrum of a loose (unconstrained) sample of complex 1 at 945.36 GHz and nominally 11 K (top, black trace) and its simulation (middle, blue trace). The simulation uses the spin Hamiltonian parameters determined from an experiment on the pellet, listed in the text. The temperature assumed in the simulation was 20 K. The bottom (red) trace is the same experimental file as at the top (black) but in which the field was adjusted according to the empirical formula Badj = Bexp – i × 8.41 μT (i being the point number). Lower panel: energy levels of a crystal of complex 1 corresponding to the same conditions as in the top panel (after the field adjustment). The arrows indicate all possible transitions for a single crystal of the S = 2 spin species regardless of their probability. Note that in the experimental spectrum, the two highest energy transitions, at 12.5 and 20.5 T, are not visible, due to their low statistical probability and low population of the levels involved
Another adjustment that was necessary to bring the simulations closer to the experimental spectrum concerned the temperature. Although the nominal temperature of the experiment shown in Fig. 7 (top) was 11 K, as measured by a Cernox® sensor located on the outside of the sample space, the actual temperature of the sample inside the holder was higher due to the heat generated by the modulation coil. This is a common problem in conventional continuous wave EPR experiments where the sample is cooled by an exchange gas rather than being in direct contact with the liquid coolant (immersion operation), in this and most cases, liquid helium, which may even be in the superfluid phase [27]. It should be noted that we observed no temperature effect on the resonance position in the experiment.
4 Conclusions
We have shown how high magnetic field-induced torquing, a ubiquitous phenomenon occurring in many paramagnetic transition metal complexes in the polycrystalline phase, can be used to an HFEPR experimentalist’s advantage provided that the alignment with the field is dependably complete and can be attributed to a particular orientation of the microcrystallites versus the field, coinciding with the crystal b axis of 1. The object of our attention was a high-spin MnIII system abbreviated as [MnLKNO3], and it served to calibrate either the frequency of certain high-frequency sources such as backward wave oscillators operating up to 1 THz, or, alternatively, the magnetic field generated by ultra-high-field resistive and hybrid magnets, reaching 36 T.
Availability of Data and Materials
The raw HFEPR files that support the findings of this study are available in the Open Science Framework repository at https://osf.io/k72xm/.
References
R.L. Carlin (ed.), Transition Metal Chemistry: A Series of Advances, vol. 3 (Marcel Dekker, New York, 1966), pp.89–201
G.E. Pake, T.L. Estle, The Physical Principles of Electron Paramagnetic Resonance (W. A. Benjamin Advanced Book Program, London, ON, Canada, 1973)
P.L.W. Tregenna-Piggott, H. Weihe, A.-L. Barra, Inorg. Chem. 42, 8504–8508 (2003)
D.P. Goldberg, J. Telser, J. Krzystek, A.G. Montalban, L.-C. Brunel, A.G.M. Barrett, B.M. Hoffman, J. Am. Chem. Soc. 119, 8722–8723 (1997)
A.-L. Barra, D. Gatteschi, R. Sessoli, G.L. Abbati, A. Cornia, A.C. Fabretti, M.G. Uytterhoeven, Angew. Chem. Int. Ed. 36, 2329–2331 (1997)
G. Aromí, J. Telser, A. Ozarowski, L.-C. Brunel, H.-M. Stoeckli-Evans, J. Krzystek, Inorg. Chem. 44, 187–196 (2005)
S.M.J. Aubin, Z. Sun, L. Pardi, J. Krzystek, K. Folting, L.-C. Brunel, A.L. Rheingold, G. Christou, D.N. Hendrickson, Inorg. Chem. 38, 5329–5340 (1999)
K.A. Campbell, M.R. Lashley, J.K. Wyatt, M.H. Nantz, R.D. Britt, J. Am. Chem. Soc. 123, 5710–5719 (2001)
Y. Sunatsuki, Y. Kishima, T. Kobayashi, T. Yamaguchi, T. Suzuki, M. Kojima, J. Krzystek, M.R. Sundberg, Chem. Commun. 47, 9149–9151 (2011)
M.C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G.L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori, R. Spagna, J. Appl. Cryst. 38, 381–388 (2005)
G.M. Sheldrick, 6.12 ed., Bruker AXS, Inc., Madison, WI (2001)
G. Sheldrick, Acta Cryst. C71, 3–8 (2015)
Rigaku and Rigaku/MSC (2000–2004). 3.6.0 ed., Molecular Structure Corporation, 9009 New Trails Dr. The Woodlands, TX 77381 USA and Tokyo 196–8666, Japan, The Woodlands, TX and Tokyo, Japan (2000–2004)
A.K. Hassan, L.A. Pardi, J. Krzystek, A. Sienkiewicz, P. Goy, M. Rohrer, L.-C. Brunel, J. Magn. Reson. 142, 300–312 (2000)
S.A. Zvyagin, J. Krzystek, P.H.M. van Loosdrecht, G. Dhalenne, A. Revcolevschi, Physica B 346–347, 1–5 (2004)
T. Dubroca, X. Wang, F. Mentink-Vigier, B. Trociewitz, M. Starck, D. Parker, M.S. Sherwin, S. Hill, J. Krzystek, J. Magn. Reson. 353, 107480 (2023)
A. Ozarowski, EPR simulation program SPIN. https://osf.io/z72tg/
J. Krzystek, G. Yeagle, J.-H. Park, M.W. Meisel, R.D. Britt, L.-C. Brunel, J. Telser, Inorg. Chem. 42, 4610–4618 (2003)
J. Krzystek, G.J. Yeagle, J.-H. Park, R.D. Britt, M.W. Meisel, L.-C. Brunel, J. Telser, Inorg. Chem. 48, 3290–3290 (2009)
J. Krzystek, S.A. Zvyagin, A. Ozarowski, S. Trofimenko, J. Telser, J. Magn. Reson. 178, 174–183 (2006)
J. Krzystek, A. Ozarowski, J. Telser, Coord. Chem. Rev. 250, 2308–2324 (2006)
P. Kottis, R. Lefebvre, J. Chem. Phys. 41, 379–393 (1964)
J. Martínez-Lillo, T.F. Mastropietro, E. Lhotel, C. Paulsen, J. Cano, G. De Munno, J. Faus, F. Lloret, M. Julve, S. Nellutla, J. Krzystek, J. Am. Chem. Soc. 135, 13737–13748 (2013)
D. Errulat, K.L.M. Harriman, D.A. Gálico, E.V. Salerno, J. van Tol, A. Mansikkamäki, M. Rouzières, S. Hill, R. Clérac, M. Murugesu, Nat. Commun. 15, 3010 (2024)
S. Stoll, A. Ozarowski, R.D. Britt, A. Angerhofer, J. Magn. Reson. 207, 158–163 (2010)
J. Krzystek, J. Telser, Dalton Trans. 45, 16751–16763 (2016)
B.M. Hoffman, V.J. DeRose, P.E. Doan, R.J. Gurbiel, A.L.P. Houseman, J. Telser, Biol. Magn. Reson. 13, 151–218 (1993)
Acknowledgements
We thank Ms. Bianca Trociewitz for her technical expertise. Dr. Yukana Kishima is acknowledged for her work as a graduate student at Okayama University.
Funding
Part of this work was performed at the NHMFL, which is funded by the National Science Foundation (Cooperative Agreement DMR 1644779 and 2128556) and the State of Florida. SH acknowledges support from the NSF (DMR-2004732).
Author information
Authors and Affiliations
Contributions
All authors contributed to the article equally.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical Approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Dubroca, T., Ozarowski, A., Sunatsuki, Y. et al. Benefitting from Magnetic Field-Induced Torquing in Terahertz EPR of a MnIII Coordination Complex. Appl Magn Reson (2024). https://doi.org/10.1007/s00723-024-01706-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00723-024-01706-3