
Abstract
Paramagnetic centers (PCs) of Ti3+ and Cr3+ were detected in polycrystalline Cr-doped TiO2 (rutile) semiconductors in which the chromium content varied from 0.1 to 1.7 at.%. For the first time, the energy position of the Cr3+ ion in the band gap of such oxide semiconductors was determined using the electron paramagnetic resonance (EPR) technique with illumination in situ. The irradiation effect was reversible. We believe that the increase of the EPR signal intensity under illumination is a result of capturing the photoexcited charge carriers by chromium ions with their subsequent transition to the state of Cr3+ paramagnetic centers (PCs) whose energy levels are located practically in the middle of the band gap, ca. 1.45 eV below the conduction band of TiO2 semiconductor. The band gap was determined by optical experiments. Spin Hamiltonian parameters (g, D, and E values) have been obtained by EPR spectra simulation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Titanium dioxide (TiO2) is one of the semiconductors that can be applied as photocatalyst, self-cleaning surface additives, and photosensitive electrodes in photoelectrochemical cells (PEC) for water electrolysis due to its high stability and resistance to photocorrosion [1, 2]. However, the large TiO2 bandgap (Eg ≥ 3.0 eV) prevents its widespread use as a material for solar energy conversion to chemical energy [3,4,5] because the absorbed UV light is only a small part (< 4%) of the solar spectrum. It is known that the photosensitivity of TiO2 in the visible region can be enlarged either by decreasing Eg or via increasing the absorption coefficient α at wavelengths λ > 450 nm. One of the most attractive approaches for this can be the synthesis of mixed oxides: doping of TiO2 with electroactive additives – various ions of transition metals. Earlier research showed [6,7,8,9] that a significant increase of α in the visible region, as a rule, does not lead to the improvement of the PEC properties of mixed oxides such as, e.g., {TiO2/MxOy} where M are various transition metals.
Doping additives have either donor (e.g., niobium) or acceptor (vanadium, chromium, iron, etc.) properties and usually doping has been carried out by metal ions of only one type in the concentration interval between 0.1 and 10 at.% [8,9,10,11]. Among transition metals, Cr3+ ions are of large interest. Indeed, Refs. [12, 13] demonstrated that doping the rutile lattice by chromium ions with up to ~ 1020 atom⋅cm−3 results in a significant increase of spectral sensitivity of electrodes up to wave lengths of 555–570 nm. Although the influence of the dopant on semiconductor and PEC features of TiO2 were investigated in detail, the analysis of the structure and spatial organization of these doped systems has not been paid enough attention to.
Nevertheless, the influence of the ligating additives on the efficiency of solar energy conversion has been studied [14], e. g. for dopants such as Re, Nb, V, Cr and for oxygen vacancies in photoelectrodes made from polycrystalline rutile. The increase in efficiency transformation of the rutile photoelectrode upon rhenium doping has been demonstrated. Doping of TiO2 by vanadium and chromium resulted in widening of the sensitivity region of electrodes in the visible region. Electrophysical and photoelectrochemical properties of polycrystalline TiO2 were studied as a function of the chromium content [15]. It was shown that increasing the concentration of chromium in rutile photoanodes leads to an increase of the visible light photosensitivity and simultaneously a higher rate of surface recombination and the anode photocurrent moves towards positive values.
Physical properties of Cr-doped polycrystalline TiO2 (rutile) such as electrical conductivity, pycnometric density and spectral dependence of the anode photocurrent as well as the short-circuit current dependence of Ti1-xCrxO2 composites have been reported [13]. A dependence of the anode photocurrent on potential and spectral dependence of the photoelectrolysis current were studied in Ref. [14].
The photocatalytic activity of chromium doped TiO2 photocatalysts prepared by different methods such as high-temperature synthesis of polycrystalline rutile-type materials [13,14,15], a one-step flame spray pyrolysis technique [16, 17], hydrothermal synthesis [18, 19], sol–gel method [20], has been studied in detail in various photochemical reactions at various conditions (temperatures, reagent concentrations, solvents etc.) [16,17,18,19,20,21,22,23,24]. It was revealed that Cr-doped TiO2 in anatase and rutile nanoparticles demonstrated extended visible light response of TiO2. The increased content of rutile and Cr3+ in the titania, has effectively promoted the formation of oxygen vacancies in the material. At contents higher than 1 at.%, Cr is forming Cr2O3 clusters and the photocatalytic activity of Cr-TiO2 photocatalytsts starts to decrease. Indeed, it was shown in many articles that at low content of chromium the photocatalytic activity of Cr-TiO2 increases in parallel with its better visible light absorption but at high Cr3+ content it dropped down significantly while the visible light absorption continued increasing.
As it was already mentioned above, many physical methods such as XRD, TEM, AFM, EPR, XPS, UV–vis diffuse reflectance, and FT-IR spectroscopy were used for studying the influence of chromium doping and other factors on the phase composition, microstructures, optical and photocatalytic properties of Cr-TiO2 semiconductors. Among these methods, electron paramagnetic resonance (EPR) is one of the most informative since chromium centers exist in the TiO2 lattice or on its surface mainly as Cr3+ and/or Cr5+ ions (electronic states are 3d3 and 3d1 respectively, the nuclear spin of the main isotope 52Cr is I = 0; the nuclear spin of the isotope 53Cr is I = 3/2, nature abundance is 9.5%) [25,26,27]. Therefore, in many articles structural results on Cr-TiO2 systems were reported using EPR techniques, e.g., in [27,28,29,30,31,32,33,34]. Such investigations were currently continued [16, 19, 35,36,37] and confirmed that the main state of chromium in Cr-TiO2 is Cr3+ and its content is close to its total amount in the crystal lattice of TiO2 when chromium concentration does not exceed 1 at.%.
In this work, the structural and photophysical properties of polycrystalline TiO2 (rutile) doped with chromium at concentrations from 0.1 to 1.7 at.% were investigated in detail using EPR spectroscopy with in-situ UV–Vis light irradiation, spectra simulation, and UV–Vis spectroscopy. Chromium ions have been chosen due to the fact that the Cr3+ ionic radius is very close to that of the Ti4+ ion which contributes to a better solubility of the dopant.
2 Experimental
Powders of TiO2 and the doping admixture of Cr2O3 both of especially pure grade were used as precursor substances. The polycrystalline Cr-TiO2 samples were synthesized by the solid-phase high-temperature reaction [15]. These two oxides were mixed in the required ratio, thoroughly stirred and formed into bricks which were thermally treated for 5 h at 1200 °C in an inert medium of He. To remove oxygen vacancies after the high-temperature annealing, the samples were additionally treated in air for 2 h at 800 °C. Due to the doping chromium atoms, the samples became dark-colored both on the surface and in the bulk. The homogeneity of the resulting samples was controlled by X-ray diffraction analysis, which confirmed that all doped materials retained uniformity and had the structure of rutile and coincided with the data published in [15].
The EPR spectra were recorded using a Varian E-3 and Bruker ELEXSYS-E500 X-band CW EPR spectrometers at 77 K in thin-walled quartz tubes, 4.0 mm in diameter. DPPH (g0 = 2.0036) and Mn2+ ions in MgO matrix were used as standards for exact calculating the magnetic field magnitude. Positions, line widths and line intensities have been measured from the experimental EPR spectra, which were processed using the computer Program package provided by Prof. A. Kh. Vorob’ev (Department of Chemistry, M. V. Lomonosov Moscow State University) [38]. The samples were illuminated directly in the spectrometer cavity in the range of 300 − 900 nm by monochromatic light. As the light source, a halogen lamp was used in the first case, and a 50 W high pressure mercury lamp equipped with the diffraction monochromator was used in the second case. The illumination intensity was approximately 40 mW·cm−2. A flat EPR sample cell of 1 mm thickness was used to provide a more uniform illumination of the samples.
Computer simulation of EPR spectra of the Cr-TiO2 samples was carried out for experimental EPR X-band spectra using the theoretical approach described in Ref. [39] for ions with the electron spin S equal to 3/2 using Belford eigenfield method [40]. The best parameters of D, E and the g-tensor components as well as the relative content of the paramagnetic centers (PCs) are discussed below.
The diffuse light reflection spectra of the TiO2 and Cr-TiO2 samples were recorded by a LS-55 Perkin Elmer (USA) spectrometer, which allows registration of the light scattering from the sample surface in a spectral range between 200 and 800 nm with spectral slit widths from 2.5 to 20 nm. The same equipment has been used for fluorescence spectra registration.
3 Results and discussion
It has been discussed in the Introduction that the region of visible light absorption in Cr-doped titania is shifting to the long-wave area (a red-shift) in parallel with the chromium content in a semiconductor, and the efficiency of such photocatalysts grows until ca. 1% of doping, and noticeably decreases at higher chromium concentration. Changes occurring in the Cr-TiO2 system upon a variation of the Cr-to-TiO2 ratio was not clarified yet. Therefore, we have employed the EPR method to study these changes in the present paper.
Figure 1 presents experimental EPR spectra at 77 K of Cr-doped TiO2 at a chromium content [Cr]0 between 0.7 and 1.7 at.%. The spectrum of pure TiO2 with Ti3+ ions is also shown for a comparison. All spectra are of complex shape mainly consisting of a rather intensive central line at a g-factor value g ≈ 1.98 (at B ≈ 344 mT) and a group of four lines with various shape and intensity at the magnetic field B equal to ca. 140, 260, 408, and 485 mT. One can see that the line of Ti3+ ions in the bulk of the TiO2 lattice is observed at a magnetic field B somewhat higher (gTi = 1.957 ± 0.003) than that of the central chromium signal. The gTi value measured in this work corresponds well to that known from the literature for polycrystalline rutile materials: 1.955 [41], 1.965 [42, 43].

Experimental EPR spectra at 77 K of Cr-doped TiO2: a—pure TiO2 enriched with Ti3+ ions, b—0.7, c—1.2, d—1.5, and e—1.7 at.% of Cr3+
The increase of the chromium concentration is reflected in a few marked changes in the EPR spectra: (i) total growth of the intensity of the Cr3+ spectra, (ii) changes of the ratio between the central chromium line and the multiplet spectrum, (iii) appearance of a new very broad line (Fig. 1, spectra d, e), and (iv) broadening of Cr3+ lines marked as * and ** in Fig. 1, which is demonstrated in Fig. 2. According to the classification of energy levels of the chromium ion given in Ref. [32], the line “*” represents a transition between energy levels of the lower doublet at the magnetic field parallel to the x-axis while the line “**” corresponds to the transition between energy levels of the upper doublet when H is parallel to the x-axis. The nature of the broad line (its width is more than 200 mT) is not really clear but we suppose that it appears due to formation of Cr2O3 microphases (clusters) in the TiO2 matrix at [Cr]0 > 1 at.% [16, 21].

Concentration broadening of the EPR linewidths ΔB for EPR lines * (1) and ** (2) as a function of the Cr3+ content in TiO2. T = 77 K
It is well known [39, 44] that such linear dependence of the individual line width as a function of concentration is observed in a solid state (or at low temperatures) due to the dipole–dipole interaction among paramagnetic centers (PCs) if their spatial distribution is random or regular by the lattice sites. In our case, both lines * and ** are not uniformly broadened and moreover, we don’t exactly know between which electron spin levels the transitions occur. Therefore, we cannot determine local concentrations of the Cr3+ PCs in the TiO2 matrix, but according to [39, 44] we can argue that the broadening of these lines is caused by the magnetic dipolar interaction.
Spin Hamiltonian parameters g-factor, D, and E were determined from the computer simulation of the experimental EPR spectra. Examples of such simulation are shown in Figs. 3 and 4 for samples with 0.7 and 1.5 at.% of chromium respectively.

Experimental (a, c; blue) and simulated (b, d, black) EPR spectra (77 K) of a Cr-TiO2 sample with 0.7 at.% of chromium. (c) and (d) are the parts of spectra (a) and (b) correspondingly, recorded at sevenfold bigger gain coefficient

Experimental (a, blue) and simulated (b, black) EPR spectra (77 K) of a Cr-TiO2 sample with 1.7 at.% of chromium
The basic states of Cr3+ ions in titania at 77 K are the orbital singlet and a spin quadruplet in the octahedral coordination sphere [39]. The cubic field does not split spin levels of the ion with the electron spin S = 3/2, thus, only one non-resolved state (and EPR line) with the isotropic g-factor value equal to ~ 1.98 should be observed (Fig. 3a, b). Crystal fields of the lower symmetry reduce the degeneracy by the spin and splitting of the quadruplet on two Kramers doublets is observed in all experimental spectra of Cr-TiO2 samples containing different chromium amount. Spectra were simulated as a set of mononuclear PCs spectra with spin S = 3/2 and various parameters of the fine structure interaction tensor (D and E). These spectra were described as a sum of weakly split signals (the central line) and of strongly split lines, i.e. PCs, whose fraction increases with increasing chromium content in the composite. Unfortunately, we were not successful to determine a set of the fine structure interaction parameters which could satisfactorily reproduce all four peaks related to the strongly split spins (S = 3/2). A theoretical spectrum of Cr2 centers, whose parameters are usually reproduced in the literature, is shown in Fig. 4. One can see that the low-field peak is a little bit shifted compared to the experimental signal, and this is the best fitting.
All spectra are described by a rhombic-distorted spin Hamiltonian Ĥ with fine structure:
where S = 3/2, Sx, Sy, Sz are projections of the full spin on axes x, y, and z, respectively; D, E are the main components of the fine structure coupling tensor; g is the isotropic value of the g-tensor; H is the external magnetic field.
The best agreement between experimental and simulated EPR spectra was achieved for D, E and the g-factor parameters listed in Table 1. Here the assumption was made that in each sample there exist all four Cr3+ paramagnetic centers with the relative concentrations presented in Table 2 for all chromium contents studied. We wish to note that (i) the g-factor is the same for all four PCs, (ii) the Cr1 PCs are related to Cr3+ ions with the orbital singlet of the non-resolved electronic state, (iii) the relative concentration of Cr1 PCs decreases more than tenfold from 0.7 at.% to 1.7 at.% of chromium content while the portions of the rest three PCs (Cr2, Cr3, Cr4) remain comparable and rather stable (Table 2). Figure 5 demonstrates the relative contributions of all four PCs to the EPR spectrum of the sample containing 1.5 at.% of chromium. Unfortunately, we still cannot attribute to which Cr3+ structures these Cr2, Cr3, and Cr4 complexes relate although it is clear that they differ by the local environment in the TiO2 lattice.

Experimental (a) and simulated (b–e) EPR spectra (77 K) of a Cr-TiO2 sample with 1.5% of chromium of PCs Cr1 (b), Cr2 (c), Cr3 (d), and Cr4 (e) (see Table 1)
When comparing parameters listed in Table 1 with those known from the literature for Cr-TiO2 systems, e.g.: g = 1.97, D = 0.68 and E = 0.14 cm−1 [28, 29]; g = 1.98, D = 0.679 and E = 0.143 cm−1 [32]; g = 1.978 [37], we would say that they are rather identical. Nevertheless, our experimental spectrum of the strongly split Cr3+ ions pattern cannot be reproduced using only one set of fine structure coupling parameters. In Ref. [29], the angular dependence of the EPR spectra is for a mono-crystal while in Ref. [32] a theoretical spectrum is not reproduced.
A necessary step of our study was the determination of the band gap energy value (Eg) of our semiconductors. The measurements were carried out by the traditional method of recording diffuse reflection optical spectra [4, 5], which are presented for our composites and bare TiO2 in Fig. 6.

Diffuse reflection optical spectra for bare TiO2 (a), TiO2 enriched with Ti3+ PCs (b), and Cr-TiO2 with 0.1 at.% (c) and 1.0 at.% (d) of chromium
Compared to the original TiO2 sample, all the annealed and Cr-doped TiO2 oxides demonstrate a weak additional absorbance in the region 370–390 nm, which is usually attributed to titanium 3d1 (Ti3+) states [45, 46].
The band gap values Eg of the samples studied were determined using the Kubelka and Munk two-component theory [47] under the assumption that the irradiating light is monochromatic and the diffusely reflected radiation is isotropic. According to this theory, diffuse reflection R of the sample depends only on the ratio of the absorption coefficient α and the scattering coefficient S, and not separately on the scattering coefficient or absorption coefficient:
where F(R) is the Kubelka–Munk function. To determine the band gap value for the direct band gap semiconductor, the experimental data may be represented as the dependence
and for the indirect band gap semiconductor, usually the experimental data may be represented as following:
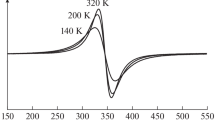
where h is the Planck constant, and ν is the radiation frequency. The band gap Eg has to be determined from the intersection point of the linear extrapolation of this dependence with the abscissa axis at α = 0. Following this approach we have determined Eg values using the Eq. (3) first. Figure 7 presents Kubelka–Munk dependences for polycrystalline bare TiO2 (rutile) and Cr-TiO2 with 0.1 at.% of chromium. Eg values calculated by this method are listed in Table 3.

Kubelka–Munk dependences for polycrystalline bare TiO2 (A) and 0.1 at.% Cr in Cr-TiO2
The band gap energy value can also be determined using Eq. (4) in the case of the indirect band gap semiconductor. The results are also given in Table 3 as well as Eg parameters measured from luminescence studies shown in Fig. 8. One can see from the data presented in Table 3 that all three approaches show very similar values of the band gap energy.

Luminescence excitation (a, c) and fluorescence spectra (b, d) of Cr-doped TiO2 at 0.1 (a, b) and 1.0 (c, d) at.% of chromium
We would note that the optical band gaps calculated using the Kubelka–Munk theory may be slightly lower than the real energy difference between the borders of the valence and conduction bands due to the rich spectrum of the surface states in the polycrystalline TiO2, especially near the borders of the band gap. We can see the evidence of this fact when comparing the data of the light scattering and the luminescence. Luminescence spectra represent sharp enough bands usually attributed to the excitons annihilation in the semiconductor grains.
The next step of our studied was concerned with the effect of illumination on the paramagnetic centers and obtaining the important characteristic of doped semiconductors such as the value of the dopant energy level in the band gap. To estimate the position of such energy levels of Cr3+ ions in the Cr-TiO2 band gap, we have studied the dependence of the EPR signal intensity of the appropriate Cr3+ paramagnetic center as a function of the irradiation light wavelength λ. The Cr1 signal was chosen due to its intensity that is much bigger compared to other lines in the EPR spectrum of the sample with 1.0 at.% of chromium. Illumination of the sample was carried out directly in the EPR spectrometer resonator, which allowed recording of the EPR spectra in situ following recommendations of Ref. [48]. After illumination at a certain wavelength before starting irradiation at the next wavelength, we had to wait for the decay of the relaxation processes of the EPR line amplitude back to that under dark conditions. This waiting time was approximately 40 min. Results of illumination at λ = 950 and λ = 750 nm are shown in Fig. 9.

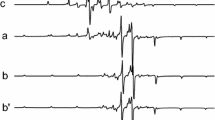
Experimental EPR spectra at 77 K of Cr-doped TiO2 with 1% of chromium under illumination with λ = 950 (a, a') and λ = 750 nm (b, b'). Spectra (a', b') are enlarged fivefold compared to (a, b)
One can see that the amplitude of the EPR spectrum components increases under illumination but the shape of the EPR signal does not change much at different wavelengths.
We have carried out further experiments for checking the dependence of the EPR signal amplitude on the wavelength of light irradiation, which is presented in the form of a diagram in Fig. 10. One can see that changing the irradiation light from long wavelengths to the visible and UV areas, a sharp increase of the central Cr3+ signal, Cr1, occurs at ca. λ = 850 nm or at an energy equal to 1.45 eV.

Dependence of the EPR signal of the Cr1 intensity on the wavelength of illumination for Cr3+ PCs in polycrystalline TiO2
The effect of light illumination is completely reversible, which reflects the process of recharging PCs in the Cr-TiO2 matrix in the cycle “dark–light–dark”. The optical width of the band gap Eg determined from optical measurements is equal to ca. 2.9 eV although this value could be somewhat smaller than the correct value of Eg. The obtained results allowed us to construct the band gap diagram of our polycrystalline material with the appropriate energy level of the dopant (defects) within the band gap (Fig. 11).

A schematic energy diagram for the Cr-TiO2 semiconductor
According to the determined value of the photon energy (1.45 eV) at which the recharging of the defects occurs, the energy levels of Cr3+ ions are localized approximately in the middle of the band gap.
To explain these results, we assume that in Cr-TiO2 samples, besides paramagnetic Cr3+ centers, there also exist some diamagnetic chromium ions, e.g. Cr4+. Under light irradiation, an electron passes from the valence band to Cr4+ center which energy level is localized in the band gap. Accepting the electron, Cr4+ center transforms to the paramagnetic Cr3+ ion and this transformation leads to an increase of the amplitude of Cr3+ in the EPR spectrum.
The Cr3+ ions are a well known doping center (substitutional defect) in TiO2 with an excitation energy around a wavelength of 800 nm. This is a very active center manifested in the photoluminescence spectra, obvious in the photoluminescence excitation spectra, electroluminescence and even in optical spectra, starting from chromium concentrations of 0.005–0.02 at.%.
4 Conclusion
Samples of polycrystalline chromium doped titania (rutile) prepared by the solid-phase high-temperature (1200 °C) reaction were investigated using diffuse reflection and fluorescent optical spectroscopy and electron paramagnetic resonance (EPR) techniques including in-situ light irradiation and EPR spectra simulation. Paramagnetic centers (PCs) of Ti3+ and Cr3+ were detected and characterized. The chromium content of the samples under study varied from 0.7 to 1.7 at.%. A sample with 0.1 at.% of Cr3+ was specially synthesized for optical measurements. Spin Hamiltonian parameters (g, D, and E values) have been obtained from EPR spectra simulation. The band gap of bare and annealed TiO2 as well as of Cr-doped TiO2 samples was determined by optical experiments and was equal ca. 2.90–2.97 eV with small variations. For the first time, the energy position of the paramagnetic Cr3+ ion in the band gap of such polycrystalline semiconductors was determined using the EPR method with illumination of samples in situ. Due to the fact that the effect of illumination is reversible, we assume that the increase of EPR signal intensity under illumination reflects the capture of photoexcited charge carriers by chromium atoms with their subsequent transition to the Cr3+state. It could be shown that the Cr3+ energy level is located practically in the middle of the band gap about 1.45 eV below the conduction band of TiO2.
References
Energy Resources through Photochemistry and Catalysis, M. Graetzel, (Ed.) (Academic Press, New York, 1983)
Photoelectrochemistry, Photocatalysis and Photoreactors, M. Schiavello (Ed.) (Reidel Publ. Co., Dordrecht, 1985)
Photocatalytic conversion of Solar energy, K.I. Zamaraev, V.N. Parmon (Eds.) (Nauka, Novosibirsk, 1991) [in Russian]
Yu.V. Pleskov, Solar Energy Conversion (A Photoelectrochemical Approach) (Springer Verlag, New York, 1990)
R. Memming, Photoelectrochemical Solar Energy Conversion (Springer, Hamburg, 2005)
J. Augustinski, J. Hinden, C. Stalder, J. Electrochem. Soc. 124, 1063 (1977)
H.P. Maruska, A.K. Ghosh, Solar Energy Mater. 1, 237 (1979)
P. Salvador, Solar Energy Mater. 2, 413 (1980)
Photochemical Conversion and Storage of Solar Energy, E. Pelizzetti, M. Schiavello (Eds.) Kluwer, Dordrecht, 1991)
V. M. Arutyunian, in: Hydrogen Energy Progress, J.C. Bolcich, T.N. Veziroglu (Eds.) (EAAH, Buenos Aires, 1998), v. 1, p. 13.
V.M. Aroutiounian, V.M. Arakelyan, G.E. Shahnazaryan, Sol. Energy 78, 581 (2005)
A.K. Ghosh, H.P. Maruska, J. Electrochem. Soc. 124, 1516 (1977)
A.G. Sarkisyan, V.M. Arutyunian, G.M. Stepanyan, A.A. Pogosyan, E.A. Khachaturyan, Electrochin. 21, 261 (1985)
V.M. Arutyunian, A.G. Sarkisyan, J.R. Panosyan, V.M. Arakelyan, A.O. Arakelyan, G.E. Shakhnazaryan, Electrochin. 17, 1471 (1981)
A.G. Sarkisyan, V.M. Arakelyan, G.M. Stepanyan, R.S. Akopyan, E.L. Ignatyan, A.L. Margaryan, Sci. Notes of Yerevan State Univeraity, No. 1(146), 79 (1981) [in Russian]
B. Tian, C. Li, J. Zhang, Chem. Eng. J. 191, 402 (2012)
K.A. Michalow, E.H. Otal, D. Burnat, G. Fortunato, H. Emerich, D. Ferri, A. Heel, T. Graule, Catal. Today 209, 47 (2013)
E.D. Jeong, P.H. Borse, J.S. Jang, J.S. Lee, O.-S. Jung, H. Chang, J.S. Jin, M.S. Won, H.G. Kim, J. Ceramic Process. Res. 9, 250 (2008)
S. Ould-Chikh, O. Proux, P. Afanasiev, L. Khrouz, M.N. Hedhili, D.H. Anjum, M. Harb, C. Geantet, J.-M. Basset, E. Puzenat, Chemsuschem 7, 1361 (2014)
R. Lopez, R. Gomez, S. Oros-Ruiz, Catal. Today 166, 159 (2011)
J. Bansal, R. Tabassum, S.K. Swami, S. Bishnoi, P. Vashishtha, G. Gupta, S.N. Sharma, A.K. Hafiz, Appl. Phys. A 126, 363 (2020)
A. Hajjaji, K. Trabelsi, A. Atyaoui, M. Gaidi, L. Bousselmi, B. Bessais, M.A. El Khakani, Nanoscale Res. Lett. 9, 543 (2014)
K.A. Rahman, T. Bak, A. Atanacio, M. Ionescu, J. Nowotny, Springer-Verlag GmbH, Springer Nature, Published online, https://doi.org/10.1007/s11581-017-2370-9 (2017)
B. Santara, K. Imakita, M. Fujii, P.K. Giri, J. Alloys & Comp. 661, 331 (2016)
H.A. Kuska, M.T. Rogers, ESR of First Row Transition Metal Complex Ions (Wiley, New York, 1968)
A. Carrington, A.D. McLachlan, Introduction to Magnetic Resonance with Applications to Chemistry and Chemical Physics (Harper & Row, New York, 1967)
S.A. Al’tshuler, B.M. Kozyrev, E. P. R. of Compounds of Intermediate Group Elements (Nauka, Moscow, 1972) [in Russian]
H.J. Gerritsen, S.E. Harrison, H.R. Lewis, J. Appl. Phys. 31, 1566 (1960)
H.J. Gerritsen, in: Paramagnetic Resonance, Proc. 1-st Intern. Conf., W. Low (ed.) (Academic Press, New York, 1963). Vol. I, pp. 3–12.
T.I. Barry, Solid State Comm. 4, 123 (1966)
V.S. Grunin, G.D. Davtyan, V.A. Ioffe, I.B. Patrina, Solid State Phys. 17, 2174 (1975). (in Russian)
S. Doeuff, M. Henry, C. Sanchez, J. Livage, J. Non-Crystal, Solids 89, 84 (1987)
A. Amorelli, J.C. Evans, C.C. Roulands, J. Chem. Soc., Faraday Trans. 1 85, 4031 (1989)
J.C. Evans, C.R. Owen, C.C. Rowlands, J. Chem. Soc., Faraday Trans. 1 85, 4039 (1989)
I.S. Pentegov, E.A. Konstantinova, Phys. Stat. Solidi 8, 1954 (2011)
F. Amano, M. Nakata, J.J.M. Vequizo, A. Yamakata, A.C.S. Appl, Energy Mater. 2, 3274 (2019)
F. La Mattina, J.G. Bednorz, S.F. Alvarado, A. Shengelaya, K.A. Müller, H. Keller, Phys. Rev. B 80, 075122 (2009)
A.Kh. Vorob’ev, N.A. Chumakova, in Nitroxides: Theory, Experiment and Applications, A.I. Kokorin (ed.) (InTech Publ., Rijeka, 2012), pp. 57–112
A. Abragam, B. Bleaney, Electron Paramagnetic Resonance of Transition Ions (Clarenton press, Oxford, 1970)
G. Belford, R.L. Belford, J.F. Burkhaven, J. Magn. Reson. 11, 251 (1973)
V.V. Antufiev, Ya.V. Vasil’ev, M.P. Votinov, O.K. Kharitonova, E.V. Kharitonov, Solid State Phys., 4, 1496 (1962) (in Russian)
P.C. Gravelle, F. Juillet, P. Mériaudeau, S.J. Teichner, Faraday Discus. Chem. Soc., No. 52, 140 (1971)
A.I. Kokorin, in Chemical Physics of Nanostructured Semiconductors, A.I. Kokorin, D.W. Bahnemann (eds.) (VSP–Brill Acad. Publ., Utrecht, Boston, 2003) p. 203–261.
Y.S. Lebedev, V.I. Muromtsev, EPR and Relaxation of the Stabilized Radicals (Khimiya, Moscow, 1971). (in Russin)
X. Chen, S. Mao, Chem. Rev. 107, 2891 (2007)
N. Laidani, P. Cheyssac, J. Perriere, J. Phys. D: Appl. Phys. 43, 485402 (2010)
W. Wedland, H. Hecht, Reflectance Spectroscopy (Interscience, New York, 1966)
E.A. Konstantinova, A.A. Minnekhanov, A.I. Kokorin, T.V. Sviridova, D.V. Sviridov, J. Phys. Chem. C 122, 10248 (2018)
Acknowledgements
The experiments were performed using the facilities of the Collective Use Center at the Moscow State University. This study was partially supported by the State assignment of Russian Federation № AAAA20-120021390044-2.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Konstantinova, E.A., Ugolkova, E.A., Zaitsev, V.B. et al. Structure and Properties of Polycrystalline TiO2-Doped with Chromium Ions Studied by EPR and Optical Methods. Appl Magn Reson 53, 717–730 (2022). https://doi.org/10.1007/s00723-021-01396-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00723-021-01396-1