
Abstract
In the present study series of new Schiff bases containing indole moiety such as N′-[(5-substituted-2-phenyl-1H-indol-3-yl)methylene]-2-oxo-2H-chromene-3-carbohydrazide and their cyclocondensation products N-[2-(5-substituted-2-phenyl-1H-indol-3-yl)-4-oxothiazolidin-3-yl]-2-oxo-2H-chromene-3-carboxamides, 3-[4-acetyl-5-(5-substituted-2-phenyl-1H-indol-3-yl)-4,5-dihydro-1,3,4-oxadiazol-2-yl]-2H-chromen-2-ones, 3-[5-(5-substituted-2-phenyl-1H-indol-3-yl)-1,3,4-oxadiazol-2-yl]-2H-chromen-2-ones, and 3-chloro-4-(5-substituted-2-phenyl-1H-indol-3-yl)-1-(2-oxo-2H-chromene-3-carbonyl)azetidin-2-ones were prepared. The structures of the newly synthesized compounds have been confirmed on the basis of their elemental analyses, IR, 1H and 13C NMR, and mass spectral studies. These compounds were screened for their antioxidant and antimicrobial activities. Some of the compounds exhibited good antioxidant and antimicrobial activity.
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Schiff bases have gained importance because of physiological and pharmacological activities associated with them. Compounds containing azomethine group (–CH=N–) in the structure are known as Schiff bases, which are usually synthesized by the condensation of primary amines and active carbonyl groups. Schiff bases are well-known for their pharmacological properties as antibacterial, antifungal, anticancer, and antiviral agents [1, 2]. Similarly, the occurrence of indole ring system in numerous biologically active molecules has been recognized which plays an important role in animal and plant kingdom. Different indole-bearing compounds possess activities such as antibacterial [3–5], antifungal [6, 7], antiviral [8–10], antimalarial [11, 12], and anti-HIV [13]. Indole compounds are very efficient antioxidants, protecting both lipids and proteins from peroxidation and influences the antioxidant efficacy in biological systems [14, 15]. In recent years, many physiological properties of melatonin have been described resulting in much attention in the development of synthetic analogues of indole [16].
Literature survey reveals that chromene is one of the privileged medicinal pharmacophores which appears as an important structural component in natural compounds and generated great attention because of their interesting biological activity. Chromenes constitute the basic backbone of various types of polyphenols and widely found in natural alkaloids, tocopherols, flavonoids, and anthocyanins. It is known that certain natural and synthetic chromene derivatives possess important biological activities such as antitumor, antivascular [17], antimicrobial [18], antioxidant [19], TNF-α inhibitor [20], antifungal [21], anticoagulant, antispasmolytic, estrogenic [22], antiviral [23], anticancer [24], anti-HIV [25], antitubercular [26], anti-inflammatory [27], herbicidal, analgesic, and anticonvulsant [28].
Antioxidants are compounds that protect cells against the damaging effect of reactive oxygen species, such as singlet oxygen, superoxide, peroxyl radicals, hydroxyl radicals, and peroxynitrite. An imbalance between antioxidants and reactive oxygen species causes cancer, ageing, atherosclerosis, ischemic injury, inflammation, and neurodegenerative diseases [29]. Therefore, inhibition of oxidative damage by supplementation with antioxidant and/or free radical scavengers might reduce the risk of these diseases [30, 31]. In the past decade, medicinal chemists, food chemists, and biologists have increasingly focused their attention on researching and testing for the new and efficient synthetic antioxidants as a protective strategy against these diseases by reducing and/or inhibiting free radical reactions.
As a result, encouraged by these pharmacological properties of indole, pyrozolopyrimidine, and tetrazolopyrimidines and guided by the observation that many times the combination of one or more heterocyclic nuclei in a molecule enhances the biological profile manifold, therefore, continuation of our interest in the synthesis of biologically active indole analogues [32–35] we have synthesized the title compounds and screened them for their antimicrobial and antioxidant activities.
Result and discussion
Chemistry
In the present work, the Schiff bases 6a–6c were prepared by the condensation of equimolar ratio of 2-oxo-2H-chromene-3-carbohydrazide (4) [36] with 2,5-disubstituted indol-3-carboxaldeydes 5a–5c [37] in ethanol at reflux temperature. The obtained Schiff bases 6a–6c on cyclization with thioglycolic acid (TGA) in the presence of anhydrous zinc chloride gave compounds 7a–7c. Compound 6 on cyclocondensation with acetic anhydride yielded compounds 8a–8c. Also compound 6 on cyclization with FeCl3 in the presence of acetic acid gave compounds 9a–9c. Similarly, compound 6 on cyclization with chloroacetyl chloride in 1,4-dioxane afforded compounds 10a–10c. The synthetic route for the synthesis of all the above compounds is depicted in Scheme 1.

The structure of the newly synthesized compounds was deduced from their 1H NMR, 13C NMR, IR, and mass spectral studies. In the 1H NMR spectrum of compound 6a, the downfield signal appeared at δ = 12.35 ppm as singlet was assigned to amide NH and another singlet at 11.62 ppm integrating for one proton assigned to the indole NH, azomethine proton resonated as singlet at 8.55 ppm, whereas the twelve aromatic protons were resonated as multiplet between 7.16 and 7.92 ppm. And the signal at 6.21 ppm was resonated as a singlet of a vinyl proton of the chromene ring. The compound 6a in its 13C NMR spectrum which exhibited the signals at δ = 182.6 and 179.2 ppm was assigned to carbonyl groups of amide and chromene system, respectively. The signal at 144.2 ppm was resonated for azomethine carbon. Compound 6a in its IR spectrum exhibited characteristic absorption bands at 3291/3230, 1688/1663, 1493, 1239, and 749 cm−1 due to NH/NH, C=O/C=O, N=CH, C–O–C, and C–Cl functions, respectively. The mass spectrum of 6a exhibited the isotopic molecular ion peaks at m/z = 441 (M+) and 443 (M++2) which successfully obeys the nitrogen rule.
In the 1H NMR spectrum of the compound 7a, the downfield signal appeared at 12.39 ppm as singlet was assigned to amide NH and another singlet at 11.41 ppm integrating for one proton is assigned to indole NH, whereas the twelve aromatic protons were resonated as multiplet between 7.19 and 7.89 ppm. The vinyl proton of the chromene ring was resonated as a singlet at 6.29 ppm, the two singlets at 5.89 and 3.95 ppm resonated due to the protons of CH and CH2 groups of thiazolidinone ring, respectively. The compound 7a in its 13C NMR spectrum which exhibited the signals at 181.5, 179.2, and 178.8 ppm was assigned to carbonyl groups of amide, thiazolidinone, and chromene carbonyl groups, respectively. The signals at 60.0 and 40.0 ppm were resonated due to CH and CH2 carbons of a thiazolidinone ring. Compound 7a in its IR spectrum exhibited characteristic absorption bands at 3389/3257, 1710/1670/1627, 1400, 1220, and 746 cm−1 due to NH/NH, C=O/C=O/C=O, C–S–C, C–O–C, and C–Cl functions, respectively. The mass spectrum of 7a exhibited the isotopic molecular ion peaks at m/z = 515 (M+) and 517 (M++2) which is in good agreement with nitrogen rule.
In the 1H NMR spectrum of the compound 8a, the downfield signal appeared at 11.52 ppm as singlet was assigned to indole NH, the twelve aromatic protons were resonated as multiplet between 7.12 and 7.79 ppm. The signal at 6.39 ppm was resonated as a singlet due to CH proton of the oxadiazole ring, the singlet at 6.25 ppm was resonated due to the vinyl proton of the chromene ring and one more singlet at 3.01 ppm resonated due to OCH3 protons. 13C NMR spectrum of 8a, the signals appeared at 180.0 and 169.5 ppm were assigned to carbon of a carbonyl group attached to oxadiazole and chromene ring, respectively, the signal at 158.4 ppm was assigned to –N–C–O– of oxadiazole ring. The signal at 40.0 ppm was resonated due to acetyl CH3 of the compound 8a. Compound 8a in its IR spectrum exhibited characteristic absorption bands at 3276, 1692/1650, 1497, 1281, and 751 cm−1 due to NH, C=O/C=O, C=N, C–O–C, and C–Cl functions, respectively. The mass spectrum of 8a exhibited the isotopic molecular ion peaks at m/z = 483 (M+) and 485 (M++2) which is in good agreement with nitrogen rule.
In the 1H NMR spectrum of the compound 9a, the downfield signal appeared at 11.65 ppm as singlet was assigned to indole NH, the twelve aromatic protons were resonated as multiplet between 6.99 and 7.91 ppm. And the signal resonated as a singlet at 6.22 ppm was assigned to the vinyl proton of the chromene ring. Compound 9a in its IR spectrum exhibited characteristic absorption bands at 3271, 1632, 1486, 1243, and 744 cm−1 due to NH, C=O, C=N, C–O–C, and C–Cl functions, respectively. The mass spectrum of 9a exhibited the isotopic molecular ion peaks at m/z = 439 (M+) and 441 (M++2) which is in good agreement with nitrogen rule.
In the 1H NMR spectrum of the compound 10a, the downfield signal appeared at 12.20 ppm as singlet was assigned to amide NH and another singlet at 11.59 ppm integrated for one proton assigned to the indole NH, the twelve aromatic protons were resonated as multiplet between 7.31 and 8.25 ppm. The singlet at 6.31 ppm was assigned to the vinyl proton of the chromene ring, the signals at 5.68 and 5.36 ppm resonated due to doublet of CH/CH protons of azetidinone ring of compound 10a. Compound 10a in its IR spectrum exhibited characteristic absorption bands at 3285/3269, 1710/1694/1603, 1237, and 763 cm−1 due to NH/NH, C=O/C=O/C=O, C–O–C, and C–Cl functions, respectively. The mass spectrum of 10a exhibited the isotopic molecular ion peaks at m/z = 517 (M+), 519 (M++2), and 521 (M++4) which is in good agreement with nitrogen rule.
Antioxidant activities
Diphenyl-2-picrylhydrazyl radical scavenging activity
Radical scavenging activity (RSA) is very important due to the deleterious role of free radicals in foods and in biological systems. Diverse methods are currently used to assess the antioxidant activity of synthesized compounds. Chemical assays are based on the ability to scavenge synthetic-free radicals, using a variety of radical-generating systems and methods for detection of the oxidation of the end points. Diphenyl-2-picrylhydrazyl (DPPH) radical scavenging method is common spectrophotometric procedure for determining the antioxidant capacities of test compounds.
DPPH has been widely used to evaluate the free radical scavenging effectiveness of various antioxidant substances. In the DPPH assay, the antioxidants were able to reduce the stable radical DPPH solution to the yellow coloured diphenyl-picrylhydrazine. The method based on the reduction of alcoholic DPPH radical solution in the presence of hydrogen or electron donating antioxidant due to the formation of the non-radical form DPPH-H is usually used as a regent to evaluate free radical scavenging activity of antioxidants [38]. DPPH is stable-free radical and accepts an electron or hydrogen radical to become a stable diamagnetic molecule [39].
With this method it is possible to determine the antiradical power of an antioxidant by measuring a decrease in the absorbance of DPPH at 517 nm. Resulting a colour change from purple to yellow, the absorbance decreased in the DPPH molecule. In the radical form, this molecule has an absorbance at 517 nm which disappears after accepting an electron or hydrogen radical from an antioxidant compound to become a stable diamagnetic molecule.
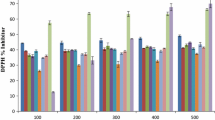
The scavenging effects of all the newly synthesized compounds 6–10 on the DPPH radical were evaluated by Hatano’s method [40]. The results were compared with the standards ascorbic acid (AA), 2-tert-butyl-4-methoxyphenol (butylated hydroxyanisole, BHA), and 2-(1,1-dimethylethyl)-1,4-benzenediol (2-tert-butylhydroquinone, TBHQ), Figs. 1, 2 and 3. The RSA results suggested that the compounds 6a, 6b, 7a, 8a, 10a, 10b, and 10c exhibited good radical scavenging activity of 84.71, 82.95, 83.45, 79.94, 84.71, 82.70, and 82.95 % at concentration 100 µg/cm3 and compounds 6c, 7a, and 10b exhibited excellent activity of 81.95, 81.70, and 81.45 % at concentration 75 µg/cm3.

Radical scavenging activity (RSA) of compounds 6 and 7

Radical scavenging activity (RSA) of compounds 8 and 9

Radical scavenging activity (RSA) of compound 10
Ferric ions (Fe3+) reducing antioxidant power
The reductive ability of synthesized compounds was assessed by the extent of conversion of Fe3+/ferricyanide complex to the Fe2+/ferrous form. The reductive powers of the compounds were observed at different concentrations and results were compared with standards BHA, TBHQ, and AA. The reducing ability of the synthesized compounds indicated that the increase in the concentration of samples increases Ferric ions reducing antioxidant power (FRAP).
The reductive ability results (Figs. 4, 5, 6) suggested that the compounds 6a, 10a, 10b, and 10c exhibited good reducing power ability at concentration 25, 100, 100, and 100 µg/cm3, respectively. These compounds reduced metal ion complex to their lower oxidation state or take part in electron transfer reaction. In other words, these compounds showed the ability of electron donor to scavenge free radicals. The rest of the test compounds showed lower absorbance as compared to the standards. The higher the absorbance of the compounds indicated greater the reducing power.

Ferric ions (Fe3+) reducing antioxidant power (FRAP) of compounds 6 and 7

Ferric ions (Fe3+) reducing antioxidant power (FRAP) of compounds 8 and 9

Ferric ions (Fe3+) reducing antioxidant power (FRAP) of compound 10
Ferrous (Fe2+) metal ion-chelating activity
Iron, in nature, can be found as either ferrous or ferric ion, with the latter form of ferric ion predominating in foods. Ferrous ions chelating may render important antioxidative effects by retarding metal-catalyzed oxidation. Ferrous ion-chelating activity of synthesized compounds is shown in Figs. 7, 8 and 9.

Ferrous (Fe2+) metal ion-chelating activity of compounds 6 and 7

Ferrous (Fe2+) metal ion-chelating activity of compounds 8 and 9

Ferrous (Fe2+) metal ion-chelating activity of compound 10
The chelating effect of ferrous ion by synthesized compounds and standards was carried out by literature method. Among the transition metals, iron is known as the most important lipid oxidation pro-oxidant due to its high reactivity. The effective ferrous ions chelators may also afford protection against oxidative damage by removing iron that may otherwise participate in HO· generating Fenton type reactions [41]:
Ferric (Fe3+) ions also produce radical from peroxides although the rate is tenfold less than that of ferrous (Fe2+) ions [41]. Ferrous ion is the pro-oxidant among the various species of metal ions [42]. Minimizing ferrous (Fe2+) ion may afford protection against oxidative damage by inhibiting production of reactive oxygen species (ROS) and lipid production. Ferrozine can quantitatively form complexes with ferrous ions in this method. In the presence of chelating agents, the complex formation is disrupted resulting in a decrease in red colour of the complex. Measurement of colour reduction, therefore, allows estimating the metal-chelating activity of the co-existing chelators [43]. Lower absorbance indicates higher metal-chelating activity. In this assay, synthesized compounds interfered with the formation of ferrous and ferrozine complex. Compounds 6a, 7a, 8a, 9a, 10a, 10b, and 10c exhibited good chelating activity. These results suggested that these compounds have the ability to capture ferrous ions before ferrozine.
Antimicrobial activity
All the newly synthesized compounds 6–10 were assessed for their in vitro antibacterial activity against four representative bacterial species, viz., Escherichia coli (MTCC-723), Staphylococcus aureus (ATCC-29513), Klebsiella pneumonia (NCTC-13368), and Pseudomonas aeruginosa (MTCC-1688) using gentamycin as reference. Determination of MIC was done using the serial dilution method [44, 45]. The results are tabulated in the Table 1.
In vitro antifungal activity of the synthesized compounds 6–10 was assessed against representative fungal species, viz., Aspergillus oryzae (MTCC-3567T), Aspergillus niger (MTCC-281), Aspergillus flavus (MTCC-1973), and Aspergillus terreus (MTCC-1782) using fluconazole as a reference, by serial dilution method [46, 47].
The minimal inhibitory concentration (MIC) values obtained by the broth microdilution method are tabulated in Table 1. Synthesized compounds have comparable and similar inhibitory effects (low to moderate MIC values 8 and 512 µg/cm3). The antibacterial activity results revealed that compounds 10a showed excellent activity with MIC 4 µg/cm3 against E. coli and K. pneumonia and rest of the compounds showed moderate activity (MIC from 8 to 512 µg/cm3) against tested bacteria, which is shown in the Table 1.
On the other hand, the antifungal activity results revealed that compound 10b exhibited excellent activity with MIC 4 µg/cm3 against A. oryzae and rest of the compounds showed moderate activity (MIC values 8 to 512 µg/cm3) against tested fungi.
Conclusions
In conclusion, the present study revealed that the compound 10 having two chlorine atoms on phenyl rings showed excellent antioxidant and antimicrobial activity. Hence, it clears that the presence of electronegative chlorine atom on phenyl ring enhances the biological activity of the synthesized compounds.
Experimental
Elemental analysis was obtained from Perkin Elmer 2400 CHN elemental analyzer, a microprocessor-based instrument. All the compounds gave C, H, and N analyses within ±0.3 %. IR spectra of the synthesized compounds were recorded as KBr pellets on a Perkin Elmer-Spectrum RX-I FT-IR instrument covering the range 4000–400 cm−1. The 1H NMR and 13C NMR spectra were recorded using DMSO-d 6 as a solvent with a Bruker NMR spectrometer at 500 and 125 MHz, respectively. The chemical shift values are expressed in ppm (δ scale) using tetramethylsilane as an internal standard. The mass spectral measurements were carried out by Waters Q-TOF MICROMASS (LC–MS).
Laboratory chemicals were supplied by Merck and Himedia Ltd. and were of high purity grade; solvents were distilled and dried before use. Melting points of the synthesized compounds were determined by electro-thermal apparatus using open capillary tubes. The purity of the compounds was checked by TLC using silica gel-G-coated aluminium plates (Merck) and spots were visualized by exposing the dry plates in iodine vapours. The precursor 2-oxo-2H-chromene-3-carbohydrazide (4) was prepared by the literature method [36] and 2,5-disubstituted indol-3-carboxaldehydes 5a–5c were prepared according to the reported method [37].
General procedure for the preparation of N′-[(5-substituted-2-phenyl-1H-indol-3-yl)methylene] 2-oxo-2H-chromene-3-carbohydrazides 6a–6c
Carbohydrazides 4 (0.01 mol) and 2,5-disubstituted indole-3-carboxaldehydes 5a–5c (0.01 mol) in ethanol were refluxed on water bath for 6–8 h, the progress of the reaction was monitored by TLC. After the completion of the reaction, the reaction mixture was decomposed in crushed ice, the separated solid product was filtered, dried, and recrystallized by ethanol. Pure product was obtained as coloured crystals.
N′-[(5-Chloro-2-phenyl-1H-indol-3-yl)methylene]-2-oxo-2H-chromene-3-carbohydrazide (6a, C25H18ClN3O3)
Pale yellow crystals; yield 0.21 g (66 %); m.p.: 242 °C; R f = 0.52 (n-hexane/ethyl acetate 2:8, v/v); 1H NMR (500 MHz, DMSO-d 6 ): δ = 12.35 (s, 1H, amide NH), 11.62 (s, 1H, indole NH), 8.55 (s, 1H, CH=N), 7.16–7.92 (m, 12H, Ar–H), 6.21 (s, 1H, vinyl CH) ppm; 13C NMR (125 MHz, DMSO-d 6 ): δ = 182.6 (amide C=O), 179.2 (chromene C=O), 144.2 (HC=N), 140.0, 135.1, 134.9, 133.7, 132.2, 130.2, 130.1, 129.8, 129.5, 127.5, 127.4, 127.2, 126.9, 125.1, 123.9, 121.4, 120.7, 119.5, 114.2, 110.1 ppm; FT-IR (KBr): \(\bar{\nu }\) = 3291, 3230 (NH/NH), 1688, 1663 (C=O/C=O), 1493 (HC=N), 1239 (C–O–C), 749 (C–Cl) cm−1; MS (70 eV): m/z = 441 (M+), 443 (M++2).
N′-[(5-Methyl-2-phenyl-1H-indol-3-yl)methylene]-2-oxo-2H-chromene-3-carbohydrazide (6b, C26H21N3O3)
Pale yellow crystals; yield 0.20 g (63 %); m.p.: 224 °C; R f = 0.55 (n-hexane/ethyl acetate 2:8, v/v); 1H NMR (500 MHz, DMSO-d 6 ): δ = 12.39 (s, 1H, amide NH), 11.60 (s, 1H, indole NH), 8.21 (s, 1H, CH=N), 7.19–8.02 (m, 12H, Ar–H), 6.22 (s, 1H, vinyl CH), 2.11 (s, 3H, CH3) ppm; 13C NMR (125 MHz, DMSO-d 6 ): δ = 181.9 (amide C=O), 178.1 (chromene C=O), 146.3 (HC=N), 140.0, 136.1, 135.2, 134.2, 133.2, 131.5, 131.0, 129.8, 129.6, 128.0, 127.7, 127.0, 127.0, 125.2, 124.1, 122.8, 121.8, 120.4, 118.3, 114.3 (Ar–C), 24.15 (CH3) ppm; FT-IR (KBr): \(\bar{\nu }\) = 3299, 3235 (NH/NH), 1698, 1660 (C=O/C=O), 1499 (HC=N), 1225 (C–O–C), 751 (C–Cl) cm−1.
2-Oxo-N′-[(2-phenyl-1H-indol-3-yl)methylene]-2H-chromene-3-carbohydrazide (6c, C25H19N3O3)
Pale yellow crystals; yield 0.22 g (65 %); m.p.: 208 °C; R f = 0.72 (n-hexane/ethyl acetate 2:8, v/v); 1H NMR (500 MHz, DMSO-d 6 ): δ = 12.40 (s, 1H, amide NH), 11.59 (s, 1H, indole NH), 8.11 (s, 1H, CH=N), 7.18–8.01 (m, 13H, Ar–H), 6.20 (s, 1H, vinyl CH) ppm; 13C NMR (125 MHz, DMSO-d 6 ): δ = 180.9 (amide C=O), 178.6 (chromene C=O), 146.8 (HC=N), 140.8, 136.7, 135.2, 134.4, 133.3, 131.7, 131.0, 129.9, 129.6, 127.9, 127.7, 127.3, 127.0, 125.3, 124.4, 122.8, 121.7, 120.5, 119.9, 114.2 (Ar–C) ppm; FT-IR (KBr): \(\bar{\nu }\) = 3285, 3240 (NH/NH), 1696, 1666 (C=O/C=O), 1492 (HC=N), 1205 (C–O–C), 755 (C–Cl) cm−1.
General procedure for the preparation of N-[2-(5-substituted-2-phenyl-1H-indol-3-yl)-4-oxothiazolidin-3-yl]-2-oxo-2H-chromene-3-carboxamides 7a–7c
Equimolar ratio of Schiff base 6 and mercaptoacetic acid were refluxed on a water bath in the presence of catalytic amount of anhydrous zinc chloride in DMF for 8–10 h. The progress of reaction was monitored by TLC, after completion of the reaction, the reaction mixture was cooled to room temperature and poured into ice-cold water. The solid separated was filtered, dried, and pure crystals of compounds 7 were obtained by the recrystallization using 1,4-dioxane as solvent.
N-[2-(5-Chloro-2-phenyl-1H-indol-3-yl)-4-oxothiazolidin-3-yl]-2-oxo-2H-chromene-3-carboxamide (7a, C27H18ClN3O4S)
Yellow crystals; yield 0.28 g (64 %); m.p.: 218 °C; R f = 0.61 (n-hexane/ethyl acetate 2:8, v/v); 1H NMR (500 MHz, DMSO-d 6 ): δ = 12.39 (s, 1H, amide NH), 11.41 (s, 1H, indole NH), 7.19–7.89 (m, 12H, Ar–H), 6.29 (s, 1H, vinyl CH), 5.89 (s, 1H, N–CH-S), 3.95 (s, 2H, S–CH2–CO) ppm; 13C NMR (125 MHz, DMSO-d 6 ): δ = 181.5 (amide C=O), 179.2 (thiazolidinone C=O), 178.8 (chromene C=O), 150.2, 139.9, 136.0, 135.2, 132.1, 130.2, 129.7, 129.5, 129.4, 129.2, 128.8, 127.6, 125.5, 124.0, 122.1, 121.0, 117.6, 114.1, 113.2, 111.3 (Ar–C), 60.0 (N–CH–S), 40.0 (S–CH2–CO) ppm; FT-IR (KBr): \(\bar{\nu }\) = 3389, 3257 (NH/NH), 1710, 1670, 1627 (C=O/C=O/C=O), 1400 (C–S–C), 1220 (C–O–C), 746 (C–Cl) cm−1; MS (70 eV): m/z = 515 (M+), 517 (M++2).
N-[2-(5-Methyl-2-phenyl-1H-indol-3-yl)-4-oxothiazolidin-3-yl]-2-oxo-2H-chromene-3-carboxamide (7b, C28H21N3O4S)
Yellow crystals; yield 0.15 g (65 %); m.p.: 235 °C; R f = 0.66 (n-hexane/ethyl acetate 2:8, v/v); 1H NMR (500 MHz, DMSO-d 6 ): δ = 12.35 (s, 1H, amide NH), 11.59 (s, 1H, indole NH), 7.20–7.91 (m, 12H, Ar–H), 6.25 (s, 1H, vinyl CH), 5.77 (s, 1H, N–CH–S), 3.92 (s, 2H, S-CH2-CO), 2.38 (s, 3H, CH3) ppm; 13C NMR (125 MHz, DMSO-d 6 ): δ = 181.0 (amide C=O), 179.2 (N–CO–CH2), 179.0 (chromene C=O), 149.3, 140.1, 138.1, 136.2, 133.2, 131.2, 130.0, 129.8, 129.2, 129.1, 129.0, 127.8, 126.2, 125.0, 123.1, 122.2, 120.1, 119.2, 116.5, 111.8 (Ar–C), 59 5 (N–CH–S), 40.1 (S–CH2–CO), 24.1 (CH3) ppm; FT-IR (KBr): \(\bar{\nu }\) = 3375, 3260 (NH/NH), 1712, 1660, 1631 (C=O/C=O/C=O), 1405 (C–S–C), 1210 (C–O–C) cm−1.
2-Oxo-N-[4-oxo-2-(2-phenyl-1H-indol-3-yl)thiazolidin-3-yl]-2H-chromene-3-carboxamide (7c, C27H19N3O4S)
Yellow crystals; yield 0.12 g (65 %); m.p.: 235 °C; R f = 0.72 (n-hexane/ethyl acetate 2:8, v/v); 1H NMR (500 MHz, DMSO-d 6 ): δ = 12.15 (s, 1H, amide NH), 11.62 (s, 1H, indole NH), 7.29-7.99 (m, 13H, Ar–H), 6.21 (s, 1H, vinyl CH), 5.59 (s, 1H, N–CH–S), 3.99 (s, 2H, S–CH2–CO) ppm; 13C NMR (125 MHz, DMSO-d 6 ): δ = 181.0 (amide C=O), 179.5 (N–CO–CH2), 179.0 (chromene C=O), 149.0, 140.2, 138.2, 136.2, 133.2, 131.2, 130.0, 129.9, 129.1, 129.0, 128.9, 127.7, 126.2, 125.0, 123.2, 122.2, 120.3, 119.2, 116.6, 111.8 (Ar–C), 60.0 (N–CH-S), 41.1 (S–CH2–CO) ppm; FT-IR (KBr): \(\bar{\nu }\) = 3320, 3275 (NH/NH), 1710, 1670, 1635 (C=O/C=O/C=O), 1402 (C–S–C), 1205 (C–O–C) cm−1.
General procedure for the preparation of 3-[4-acetyl-5-(5-substituted-2-phenyl-1H-indol-3-yl)-4,5-dihydro-1,3,4-oxadiazol-2-yl]-2H-chromen-2-ones 8a–8c
Equimolar ratio of Schiff base 6 (0.001 mol) and acetic anhydride (0.001 mol) were refluxed on an oil bath at 140 °C for 3–4 h. The reaction mixture was cooled to room temperature and poured into ice-cold water. The solid separated was filtered, washed with water, dried and pure crystals of compounds 8 were obtained by recrystallization using 1,4-dioxane as solvent.
3-[4-Acetyl-5-(5-chloro-2-phenyl-1H-indol-3-yl)-4,5-dihydro-1,3,4-oxadiazol-2-yl]-2H-chromen-2-one (8a, C27H18ClN3O4)
Dark brown crystals; yield 0.27 g (63 %); m.p.: 221 °C; R f = 0.52 (n-hexane/ethyl acetate 2:8, v/v); 1H NMR (500 MHz, DMSO-d 6 ): δ = 11.52 (s, 1H, indole NH), 7.12–7.79 (m, 12H, Ar–H), 6.39 (s, 1H, N–CH–O), 6.25 (s, 1H, vinyl CH), 3.01 (s, 3H, CH3) ppm; 13C NMR (125 MHz, DMSO-d 6 ): δ = 180.0 (N–CO–CH3), 169.5 (chromene C=O), 158.4, 147.2, 139.2, 136.0, 130.8, 130.6, 130.4, 130.2, 130.0, 129.9, 129.5, 128.1, 127.2, 126.9, 123.9, 120.6, 117.4, 115.1, 114.3, 109.7, 105.1 (Ar–C), 75.5 (N–CH–O), 23.8 (CH3) ppm; FT-IR (KBr): \(\bar{\nu }\) = 3276 (NH), 1692, 1650 (C=O/C=O), 1497 (C=N), 1281, 1220 (C–O–C/C–O–C), 751 (C–Cl) cm−1; MS (70 eV): m/z = 483 (M+), 485 (M++2).
3-[4-Acetyl-5-(5-methyl-2-phenyl-1H-indol-3-yl)-4,5-dihydro-1,3,4-oxadiazol-2-yl]-2H-chromen-2-one (8b, C28H21N3O4)
Dark brown crystals; yield 0.24 g (56 %); m.p.: 192 °C; R f = 0.55 (n-hexane/ethyl acetate 2:8, v/v); 1H NMR (500 MHz, DMSO-d 6 ): δ = 11.61 (s, 1H, indole NH), 7.19–7.98 (m, 12H, Ar–H), 6.42 (s, 1H, N–CH–O), 6.19 (s, 1H, vinyl CH), 2.99 (s, 3H, CH3), 2.04 (s, 3H, CH3) ppm; 13C NMR (125 MHz, DMSO-d 6 ): δ = 180.1 (N–CO–CH3), 172.1 (chromene C=O), 154.2, 145.1, 139.2, 136.1, 131.2, 131.0, 130.5, 130.3, 130.0, 129.8, 129.6, 128.1, 127.9, 127.0, 124.1, 122.8, 119.1, 117.1, 115.4, 111.5, 105.2 (Ar–C), 75.6 (N–CH–O), 23.2 (CH3), 22.0 (CH3) ppm; FT-IR (KBr): \(\bar{\nu }\) = 3286 (NH), 1672, 1660 (C=O/C=O), 1499 (C=N), 1242, 1222 (C–O–C/C–O–C) cm−1.
3-[4-Acetyl-5-(2-phenyl-1H-indol-3-yl)-4,5-dihydro-1,3,4-oxadiazol-2-yl]-2H-chromen-2-one (8c, C27H19N3O4)
Dark brown crystals; yield 0.27 g (62 %); m.p.: 198 °C; R f = 0.68 (n-hexane/ethyl acetate 2:8, v/v); 1H NMR (500 MHz, DMSO-d 6 ): δ = 11.72 (s, 1H, indole NH), 7.20–7.99 (m, 13H, Ar–H), 6.50 (s, 1H, N–CH–O), 6.14 (s, 1H, vinyl CH), 2.44 (s, 3H, CH3) ppm; 13C NMR (125 MHz, DMSO-d 6 ): δ = 180.1 (N–CO–CH3), 173.2 (chromene C=O), 150.2, 144.1, 138.2, 137.2, 132.1, 131.1, 131.0, 130.4, 130.3, 129.9, 129.8, 128.8, 128.1, 127.1, 125.2, 123.2, 120.2, 119.0, 116.6, 114.6, 111.4 (Ar–C), 75.5 (N–CH–O), 23.4 (CH3) ppm; FT-IR (KBr): \(\bar{\nu }\) = 3282 (NH), 1670, 1658 (C=O/C=O), 1500 (C=N), 1235, 1220 (C–O–C/C–O–C) cm−1.
General procedure for the preparation of 3-[5-(5-substituted-2-phenyl-1H-indol-3-yl)-1,3,4-oxadiazol-2-yl]-2H-chromen-2-ones 9a–9c
To a well stirred solution of compound 6 (0.001 mol) in 10 cm3 acetic acid, a solution of 1.5 g ferric chloride in 15 cm3 water was added. The reaction mixture was stirred for 2 h, diluted with 100 cm3 water, and the mixture was kept at room temperature over a night. The solid separated was filtered, washed with water, dried and pure crystals of compounds 9 were obtained by the recrystallization by using 1,4-dioxane.
3-[5-(5-Chloro-2-phenyl-1H-indol-3-yl)-1,3,4-oxadiazol-2-yl]-2H-chromen-2-one (9a, C25H14ClN3O3)
Brown crystals; yield 0.26 g (67 %); m.p.: 186 °C; R f = 0.71 (n-hexane/ethyl acetate 2:8, v/v); 1H NMR (500 MHz, DMSO-d 6 ): δ = 11.65 (s, 1H, indole NH), 6.99–7.91 (m, 12H, Ar–H), 6.22 (s, 1H, vinyl CH) ppm; 13C NMR (125 MHz, DMSO-d 6 ): δ = 181.0 (C=O), 160.2, 158.2 (O–C=N/O–C=N), 147.1, 139.3, 135.9, 130.8, 130.7, 130.5, 130.2, 130.1, 129.5, 127.2, 126.9, 123.9, 120.6, 117.4, 115.1, 114.3, 109.7, 106.2, 105.2, 97.4, 95.1 (Ar–C) ppm; FT-IR (KBr): \(\bar{\nu }\) = 3271 (NH), 1632 (C=O), 1486 (C=N), 1243 (C–O–C), 745 (C–Cl) cm−1; MS (70 eV): m/z = 439 (M+), 441 (M++2).
3-[5-(5-Methyl-2-phenyl-1H-indol-3-yl)-1,3,4-oxadiazol-2-yl]-2H-chromen-2-one (9b, C26H17N3O3)
Brown crystals; yield 0.24 g (62 %); m.p.: 192 °C; R f = 0.69 (n-hexane/ethyl acetate 2:8, v/v); 1H NMR (500 MHz, DMSO-d 6 ): δ = 11.69 (s, 1H, indole NH), 7.11–7.93 (m, 12H, Ar–H), 6.05 (s, 1H, vinyl CH), 2.04 (s, 3H, CH3) ppm; 13C NMR (125 MHz, DMSO-d 6 ): δ = 181.2 (C=O), 160.1, 158.2 (O–C=N/O–C=N), 147.0, 140.0, 136.0, 131.1, 131.0, 130.7, 130.4, 130.1, 129.5, 128.1, 127.1, 124.1, 122.6, 119.2, 117.5, 115.2, 110.4, 107.7, 105.2, 99.3, 98.1 (Ar–C) ppm; FT-IR (KBr): \(\bar{\nu }\) = 3268 (NH), 1640 (C=O), 1492 (C=N), 1221 (C–O–C), 749 (C–Cl) cm−1.
3-[5-(2-Phenyl-1H-indol-3-yl)-1,3,4-oxadiazol-2-yl]-2H-chromen-2-one (9c, C25H15N3O3)
Brown crystals; yield 0.25 g (65 %); m.p.: 172 °C; R f = 0.77 (n-hexane/ethyl acetate 2:8, v/v); 1H NMR (500 MHz, DMSO-d 6 ): δ = 11.78 (s, 1H, indole NH), 7.18–8.01 (m, 13H, Ar–H), 6.15 (s, 1H, vinyl CH) ppm; 13C NMR (125 MHz, DMSO-d 6 ): δ = 181.0 (C=O), 160.3, 159.2 (O–C=N/O–C=N), 146.9, 140.1, 136.1, 131.5, 130.9, 130.7, 130.4, 130.2, 130.0, 128.6, 127.9, 124.3, 122.8, 119.9, 117.5, 115.5, 110.0, 108.0, 105.9, 100.1, 100.0 (Ar–C) ppm; FT-IR (KBr): \(\bar{\nu }\) = 3298 (NH), 1666 (C=O), 1505 (C=N), 1210 (C–O–C) cm−1.
General procedure for the preparation of N-[3-chloro-2-(5-substituted-2-phenyl-1H-indol-3-yl)-4-oxoazetidin-1-yl]-2-oxo-2H-chromene-3-carboxamides 10a–10c
To Schiff base 6 (0.02 mol) in 30 cm3 dry benzene, few drops of triethylamine and chloroacetylchloride (0.02 mol) were added by stirring at room temperature during 15 min. The mixture was then refluxed for 1–2 h on water bath, the ethylamine hydrochloride formed was filtered off and washed several times with benzene. The filtrate and washings were combined and concentrated under reduced pressure and the residue obtained was washed with petroleum ether (40:60) to remove unreacted Schiff base. The product 10 obtained was dried and recrystallized from 1,4-dioxane.
N-[3-Chloro-2-(5-chloro-2-phenyl-1H-indol-3-yl)-4-oxoazetidin-1-yl]-2-oxo-2H-chromene-3-carboxamide (10a, C27H17Cl2N3O4)
Yellow crystals; yield 0.27 g (63 %); m.p.: 221 °C; R f = 0.58 (n-hexane/ethyl acetate 2:8, v/v); 1H NMR (500 MHz, DMSO-d 6 ): δ = 12.20 (s, 1H, amide NH), 11.59 (s, 1H, indole NH), 7.31–8.25 (m, 12H, Ar–H), 6.31 (s, 1H, vinyl CH), 5.68 (d, 1H, CH), 5.36 (d, 1H, CH) ppm; 13C NMR (125 MHz, DMSO-d 6 ): δ = 181.4, 170.5, 166.7 (C=O/C=O/C=O), 158.4, 147.2, 139.2, 135.9, 130.8, 130.6, 130.3, 130.2, 130.1, 129.5, 127.2, 126.8, 123.9, 120.6, 117.4, 115.9, 114.2, 109.7, 105.2 (Ar–C), 64.4 (azetidin C–Cl), 53.2 (azetidin C–N) ppm; FT-IR (KBr): \(\bar{\nu }\) = 3285, 3269 (NH/NH), 1710, 1694, 1603 (C=O/C=O/C=O), 1237 (C–O–C), 763, 733 (C–Cl/C–Cl) cm−1; MS (70 eV): m/z = 517 (M+), 519 (M++2), 521 (M++4).
N-[3-Chloro-2-(5-methyl-2-phenyl-1H-indol-3-yl)-4-oxoazetidin-1-yl]-2-oxo-2H-chromene-3-carboxamide (10b, C28H20ClN3O4)
Yellow crystals; yield 0.46 g (53 %); m.p.: 131 °C; R f = 0.63 (n-hexane/ethyl acetate 2:8, v/v); 1H NMR (500 MHz, DMSO-d 6 ): δ = 12.15 (s, 1H, amide NH), 11.71 (s, 1H, indole NH), 7.29–8.18 (m, 12H, Ar–H), 6.25 (s, 1H, vinyl CH), 5.66 (d, 1H, CH), 5.25 (d, 1H, CH), 2.13 (s, 3H, CH3) ppm; 13C NMR (125 MHz, DMSO-d 6 ): δ = 180.2, 172.3, 167.8 (C=O/C=O/C=O), 156.1, 148.0, 139.6, 136.1, 131.1, 130.9, 130.7, 130.4, 130.4, 130.0, 127.8, 126.5, 124.0, 121.1, 119.3, 118.0, 115.2, 111.5, 109.2 (Ar–C), 65.0 (azetidin C–Cl), 54.1 (azetidin C–N), 22.6 (CH3) ppm; FT-IR (KBr): \(\bar{\nu }\) = 3292, 3275 (NH/NH), 1718, 1686, 1620 (C=O/C=O/C=O), 1225 (C–O–C), 755 (C–Cl) cm−1.
N-[3-Chloro-2-oxo-4-(2-phenyl-1H-indol-3-yl)azetidin-1-yl]-2-oxo-2H-chromene-3-carboxamide (10c, C27H18ClN3O4)
Yellow crystals; yield 0.13 g (57 %); m.p.: 124 °C; R f = 0.78 (n-hexane/ethyl acetate 2:8, v/v); 1H NMR (500 MHz, DMSO-d 6 ): δ = 12.28 (s, 1H, amide NH), 11.75 (s, 1H, indole NH), 7.26–8.16 (m, 13H, Ar–H), 6.22 (s, 1H, vinyl CH), 5.64 (d, 1H, CH), 5.28 (d, 1H, CH) ppm; 13C NMR (125 MHz, DMSO-d 6 ): δ = 180.5, 173.1, 167.8 (C=O/C=O/C=O), 157.2, 148.1, 139.4, 136.5, 131.5, 131.0, 130.9, 130.4, 130.2, 130.0, 127.7, 126.9, 124.6, 121.4, 119.3, 118.2, 116.1, 114.4, 110.2 (Ar–C), 65.6 (azetidin C–Cl), 54.9 (azetidin C–N) ppm; FT-IR (KBr): \(\bar{\nu }\) = 3288, 3270 (NH/NH), 1710, 1678, 1628 (C=O/C=O/C=O), 1218 (C–O–C), 748 (C–Cl) cm−1.
1,1-Diphenyl-2-picrylhydrazyl (DPPH) radical scavenging activity (RSA)
The free radical scavenging activity (RSA) of compounds 6–10 at concentration (25, 50, 75, and 100 µg/cm3) was carried out in the presence of freshly prepared solution of stable-free radical DPPH (0.04 % w/v) following Hatano’s method [48], using ascorbic acid (AA), 2-tert-butyl-4-methoxyphenol (butylated hydroxyanisole, BHA), and 2-(1,1-dimethylethyl)-1,4-benzenediol (2-tert-butylhydroquinone, TBHQ) as standards. All the test analyses were performed on three replicates and the results are averaged. The result in percentage is expressed as the ratio of absorption decrease of DPPH in the presence test of compounds and absorption of DPPH in the absence of test compounds at 517 nm on ELICO SL 171 Mini Spec spectrophotometer. The percentage scavenging activity of the DPPH-free radical was measured using the following equation and the results are shown in Figs. 1, 2 and 3.
Reducing power assay
The reducing power of the synthesized compounds 6–10 was determined according to the literature method [38]. Different concentrations of samples (25, 50, 75, and 100 µg/cm3) in 1 cm3 DMSO were mixed with 2.5 cm3 phosphate buffer (0.2 M, pH = 6.6) and 2.5 cm3 potassium ferricyanide (1 %). The mixture was incubated at 50 °C for 20 min, after which a portion of trichloroacetic acid (2.5 cm3, 10 %) was added to the mixture and centrifuged for 10 min at 1000 g. The upper layer of solution (2.5 cm3) was mixed with 2.5 cm3 distilled water and 0.5 cm3 ferric chloride (0.1 %). Then absorbance at 700 nm was measured in spectrophotometer. Higher absorbance of the reaction mixture indicated greater reducing power. The results are shown in Figs. 4, 5 and 6.
Ferrous (Fe2+) metal ion-chelating activity
The chelating activity of ferrous ions by synthesized compounds 6–10 was estimated by following reported method [49]. The test samples (25, 50, 75, and 100 µg/cm3) in 0.4 cm3 ethanolic solution were added to a solution of 0.05 cm3 FeCl2 (2 mM). The reaction was initiated by the addition of 0.2 cm3 ferrozine (5 mM) and the total volume was adjusted to 4 cm3 with ethanol. Ferrozine reacted with the divalent iron forming stable magenta complex species that were very soluble in water. The mixture was shaken vigorously and kept at room temperature for 10 min. Then the absorbance of the solution was measured spectrophotometrically at 562 nm. All test analyses were run in triplicate and averaged. The percentage of inhibition of the ferrozine Fe2+ complex formations was calculated using the formula:
The control contains FeCl2 and ferrozine, complex formation molecule. The results are shown in Figs. 7, 8 and 9.
Antimicrobial activity
All the newly synthesized compounds were assessed for their in vitro antibacterial activity against four representative bacterial species, viz., Escherichia coli (MTCC-723), Staphylococcus aureus (ATCC-29513), Klebsiella pneumonia (NCTC-13368), and Pseudomonas aeruginosa (MTCC-1688) using gentamycin as reference. Determination of MIC was done using the serial dilution method [44, 45]. The materials used were 96-well plates, suspension of microorganism (0.5 McFarland), Muller-Hinton broth (Himedia) and stock solutions of each substance to be tested (2048 µg/cm3 in DMSO). The following concentrations of the substances to be tested were obtained in the 96-well plates: 1024, 512, 128, 64, 32, 16, 8, 4, and 2 µg/cm3. After incubation at 37 °C for 18–24 h, the MIC for each tested substance was determined by Bio-Rad Elisa reader (micro plate reader S/N 12883). The results are tabulated in Table 1.
References
Wang M, Wang LF, Li YZ, Li QX, Xu ZD, Qu DM (2001) Trans Met Chem 26:307
Yadav LDS, Singh S (2001) Indian J Chem 40B:440
Yamamoto Y, Kurazono M (2007) Bioorg Med Chem Lett 17:1626
Mahboobi S, Eichhorn E, Popp A, Sellmer A, Elz S, Mollmann U (2006) Eur J Med Chem 41:176
Ryu CK, Lee JY, Park RE, Ma MY, Nho JH (2007) Bioorg Med Chem Lett 17:127
Tiwari RK, Verma AK, Chillar AK (2006) Bioorg Med Chem 14:2747
Williams JD, Drach JC, Townsend LB (2005) Nucleosides Nucleotides Nucleic Acids 24:1613
Chen JJ, Wei Y, Williums JD, Drach JC, Townsend LB (2005) Nucleosides Nucleotides Nucleic Acids 24:1417
Chai H, Zhao Y, Gong CP (2006) Bioorg Med Chem 14:911
Agarwal A, Srivastav K, Puri SK, Chauhan PM (2005) Bioorg Med Chem Lett 15:3133
Kgokong JL, Smith PP, Matasabisa GM (2005) Bioorg Med Chem 13:2935
Suzen S, Buyunkbingal B (1998) Farmaco 53:525
De Martino G, La Regina G, Ragno R (2006) Antiviral Chem Chemother 17:59
Dannhardt G, Kiefer W (2001) Eur J Med Chem 36:109
Brown DW, Graupner PR, Sainsbury M, Shertzer HG (1991) Tetrahedron 47:4383
Suzen S, Alagoz Z, Puskullu MO (2000) FABAD J Pharm Sci 25:113
Gourdeau H, Leblond L, Hamelin B, Desputeau C, Dong K, Kianicka I, Custeau D, Boudreau C, Geerts L, Cai S-X, Drewe J, Labrecque D, Kasibhatla S, Tseng B (2004) Mol Cancer Ther 3:1375
Chetan BS, Nimesh MS, Manish PP, Ranjan GP (2012) J Serb Chem Soc 77:1
Mladenović M, Mihailović M, Bogojević D, Matić S, Nićiforović N, Mihailović V, Vuković N, Sukdolak S, Solujić S (2011) Int J Mol Sci 12:2822
Cheng JF, Ishikawa A, Ono Y, Thomas A, Alex N (2003) Bioorg Med Chem Lett 13:3647
Suresh T, Arunima V, Atin K, Sandeep G, Prarthana VR, Ganesh RK (2010) Acta Pol Pharm 67:423
Nareshkumar J, Jiayi X, Ramesh MK, Fuyong D, Guo J-Z, Emmanuel P (2009) J Med Chem 52:7544
Mori J, Iwashima M, Takeuchi M, Saito H (2006) Chem Pharm Bull 54:391
Aliaa MK, Manal MK, Abd El-all E, Heba AHE (2012) Int J Pharm Res Dev 4:310
Denish CK, Hetal KP, Nilesh KG (2012) Asian J Biochem Pharm Res 2:126
Nimesh RK, Dhaval DH, Prashant TM, Saurabh KP (2011) Med Chem Res 20:854
Nitin K, Sushil K, Himanshu G, Sharma PK (2012) World Res J Biochem 1:1
Bhat MA, Siddiqui N, Khan SA (2008) Acta Pol Pharm 65:235
Halliwell B (1999) Free Radical Res 31:261
Babizhayev MA, Deyev AI, Yermakovea VN, Brikman IV (2004) J Bours Drugs 5:125
Liu L, Meydani M (2002) Nutr Rev 60:368
Saundane AR, Kirankumar NM, Annapurna H, Prabhaker W (2014) J Appl Chem 3:117
Saundane AR, Prabhaker W (2012) Indian J Chem 51B:1593
Saundane AR, Yarlakatti M, Prabhaker W, Katkar V (2012) J Chem Sci 124:469
Saundane AR, Katkar V, Vaijinath AV, Prabhaker W (2013) Med Chem Res 22:806
Ramesh GCK, Yadav DB, Venkatesh KB (2010) Indian J Chem 49B:1151
Hiremath SP, Biradar JS, Purohit MG (1982) Indian J Chem 21B:249
Oyaizu M (1986) Jpn Nutr 44:307
Soares JR, Dins TCP, Cunha AP, Almeida LM (1997) Free Radical Res 26:469
Calis I, Hosny M, Khalifa T, Nishibe S (1993) Phytochem 33:1453
Miller DD (1996) Mineral. In: Fennema OR (ed) Food chemistry. Marcel Deckker, New York, p 618
Halliwel B, Gutteridge JMC (1984) Biochem J 219:1
Elmastas M, Turkekul I, Ozturk L, Gulcin I, Isildk O, Aboul-Enein HY (2006) Comb Chem High Throughput Screen 6:443
Barry AL (1980) Procedure for testing antimicrobial agents in agar media. In: Corian VL (ed), Antibiotics in laboratory medicine. Williams and Wikins, Baltimore
James D, Mac L, Jaqua JM, Sally TS (1970) Appl Microbiol 20:46
Arthington-Skaggs BA, Molestey M, Warnock DW, Morrison CJ (2000) J Clin Microbial 38:2254
Verma RS, Khan ZK, Sing AP (1998) Antimicrobial agents: past, present and future prospects. National Academy of Chemistry and Biology, Lucknow, p 55
Hatano T, Kagawa H, Yasuhara T, Okuda T (1988) Chem Pharm Bull 36:2090
Dinis TCP, Maderia VMC, Almeida LM (1994) Arch Biochem Biophys 315:161
Acknowledgement
The authors are thankful to the Chairman, Department of Chemistry, Gulbarga University, Gulbarga, for providing laboratory facilities, Chairman, Department of Microbiology and Biotechnology Gulbarga University, Gulbarga, for providing laboratory facilities to carry out antimicrobial activity, also thankful to the Director of Indian Institute of technology, Chennai and SAIF Chandigarh for providing 1H NMR, 13C NMR, and mass spectral data. Chairman, Department of Material science, for providing IR spectral data.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Saundane, A.R., Nandibeoor Mathada, K. Synthesis, characterization, and biological evaluation of Schiff bases containing indole moiety and their derivatives. Monatsh Chem 146, 1751–1761 (2015). https://doi.org/10.1007/s00706-015-1440-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-015-1440-9