
Abstract
The genome sequence of a novel circular DNA virus related to members of the genus Badnavirus was identified in diseased jujube trees by high-throughput sequencing and verified by conventional Sanger sequencing of cloned PCR products. The name "jujube badnavirus WS" (JuBWS) is proposed for this virus. Diseased jujube leaves showed yellow mosaic and malformation symptoms, with round chlorotic spots found on diseased fruit. The genome of this virus has a length of 6450 nt and has a typical badnavirus genomic structure with three open reading frames (ORFs). JuBWS was identified as a novel badnavirus based on nucleotide differences in the RNase (RT + RNase H) coding region of ORF3. The JuBWS sequence showed 70.48–76.41% nucleotide sequence identity to other known badnaviruses, thus meeting the taxonomic criterion for establishing a new species within the genus Badnavirus. This study suggested that the novel badnavirus might be a pathogen associated with jujube mosaic disease, and this will be investigated in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Chinese jujube (Ziziphus jujuba Mill.), a fruit tree of the family Rhamnaceae, is one of the oldest cultivated fruit trees in the world and is widely cultivated in China [1]. We observed mosaic and malformation symptoms of diseased leaves that were highly similar to those of leaves infected by jujube mosaic-associated virus (JuMaV) [2] in a jujube orchard in Wensu County, Xinjiang, China. Moreover, the infected fruit also had round chlorotic spots. This disease can cause jujube yield reductions of 5-80%. Therefore, we concluded that the Chinese jujube tree might be infected by a badna-like virus.
Members of the genus Badnavirus of the family Caulimoviridae infect a wide range of economically important crops worldwide. The economic losses caused by different badnaviruses in different crops range from 10% to 90% [3, 4]. Most badnaviruses infect tropical and subtropical crops [5,6,7,8,9,10]. Plants in temperate climates can also be infected by some badnaviruses [11,12,13]. The virus particles of badnaviruses are approximately 30 nm in diameter and vary in length from 120 to 150 nm, depending on the species [14]. The badnavirus genome is a circular double-stranded DNA molecule with a length of 6.9 to 9.2 kb [15]. Badnaviral genomes typically contain three open reading frames (ORFs), but they may have one or more additional ORFs. All ORFs are encoded on the sense strand of the virus [14]. Badnaviruses are spread by vegetative propagation, mealybug vectors, and, in some cases, seeds [16]. Like other members of the family Caulimoviridae, badnaviruses use reverse transcription for their replication. During replication, viral sequences are integrated into the host genome and are infectious. Symptoms of viral infection appear to be heritable as Mendelian traits and to be transmitted vertically [14].
In this study, the genome of a potentially novel badnavirus was sequenced. In July 2020, three leaf samples (WS-1, WS-2, and WS-3) were collected from a symptomatic jujube tree of the variety ‘Huizao’ grown in Wensu County and stored at −80 °C. WS-2 was used for high-throughput sequencing (HTS), WS-1 was used for virus identification and sequencing, and WS-3 was used for further analysis. The leaf samples showed mosaic and malformation symptoms (Supplementary Fig. S1B and C). Leaves from asymptomatic trees were used as controls.
The sequencing platform was an IlluminaHiSeq XTen high-throughput sequencer with a paired-end 150-bp setup (Biomarker Biology Technology Ltd. Company, Beijing, China). The raw reads were trimmed to remove adaptor sequences and filtered to remove low-quality reads, using FASTP version 1.5.6. The trimmed sequence reads were spliced into longer contigs using Velvet version 1.2.08 [17] and IDBA-UD version 1.1.1 [18]. A total of 44,533,809 raw SRNA reads were obtained, and 39,607,094 clean reads were assembled de novo using Velvet after removing adaptor sequences and filtering for quality. The resulting contigs were used to search in the nr and nt databases of NCBI (http://www.ncbi.nlm.nih.gov/) using BLASTx and BLASTn. A total of 61 contigs with lengths between 47 and 448 nt were found in the samples; these sequences showed similarity (between 41.79% and 93.33% identity) to several badnaviruses. Fifty-six contigs ranging in length from 47 to 464 nt exhibited 73.17%-100% sequence identity to Jujube Badnavirus BJ (MN274946, unpublished).
Total DNA was extracted from WS-1 leaf samples that were obviously symptomatic, using a DNA Secure Plant Kit. DNA fragments were amplified by PCR, using four specific primer pairs, purified, and sequenced to determine the complete genome sequence of the virus. In addition, a specific primer pair, FP1/RP1, was designed based on the HTS results to amplify a coding region that is conserved in badnaviruses (Supplementary Table S1). We collected and tested diseased leaf samples from six sites in the Aksu region with the same symptoms (Supplementary Fig. S2). PCR amplifications were performed as follows: The reaction mixture (25 µL) contained 1 µl of DNA template, 13 µl of 2× Es Taq Master Mix (CWBIO), 1 mM each primer, and 9 µl of ddH2O. PCR cycling was performed as reported previously [2]. The annealing temperature and extension time were adjusted to fit the primer combination used in each reaction and the size of the expected PCR product. The PCR products were separated by 1.5% agarose gel electrophoresis, stained with ethidium bromide, and viewed under ultraviolet light. PCR products were extracted from the gel and purified using an EasyPure® Quick Gel Extraction Kit and cloned into the pEASY-T1 cloning vector. At least three positive clones of each PCR product were sequenced in both directions at Sangon Biotech (Shanghai) Co., Ltd. All three clones of each fragment showed over 99.9% sequence identity. The genome sequences obtained were assembled into continuous sequences based on overlapping common regions (>100 bp) using SnapGene® Viewer version 4.3.6. The sequence of a circular DNA molecule of 6450 bp was obtained, and this was considered to represent the genome of a possible novel virus, referred to as "jujube badnavirus WS" (JuBWS). The full-length sequence of the circular DNA virus has been deposited in the GenBank database with the accession number OL739567.
The edited sequence was identified by a BLAST search of the NCBI GenBank database (http://www.ncbi.nlm.nih.gov/genbank/), and NCBI ORFfinder was used to predict ORFs with a minimum length of 75 nt (https://www.ncbi.nlm.nih.gov/orffinder/). A genome map was generated using SnapGene® Viewer version 4.3.6. Conserved domains of hypothetical gene products were identified using the NCBI conserved domain tool (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi).
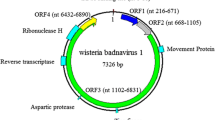
Analysis of the genome and conserved domains showed that the genome structure of JuBWS is typical of members of the genus Badnavirus (Fig. 1A, right panel). The JuBWS genome contains a tRNAMet binding site (TGGTATCAGAGC1–12), which begins at the 5’ end of the viral plus strand as the primer for minus-strand synthesis [3]. A TATA box (TATAAAT6291–6297) and a CAAT box (GCCCAAT6246-6252) are present in the noncoding intergenic region upstream of the tRNAMet site. The genome contains three typical badnavirus open reading frames (ORFs), all on the plus strand (Fig. 1A, left panel). An uncharacterized superfamily domain approximately 200 residues long in ORF1, referred to as ‘domain of unknown function 1319’ (DUF1319) [19], is restricted to badnaviruses. No conserved domains were identified in ORF2. ORF3 of JuBWS contains four domains: a zinc-finger domain, a pepsin-like aspartate protease domain, an RT domain, and an RNase H domain. The coding region of the JuBWS movement protein (MP) is at the 5' end of ORF3. The RNase coding region within ORF3 is the most conserved region of the genome. The nucleotide (nt) sequence identity of this region is less than 80%, allowing this region to be used to distinguish species in this genus [14]. JuBWS is most similar to JuMaV, with 73.22% sequence identity and shows 76.41% identity to JuMaV in the RNase region. Jujube badnavirus BJ (MN274946, unpublished) shares 99.6% identity with JuBWS, and these are possibly different strains of the same virus. The RNase region of JuBWS shares 70.48-76.41% nucleotide sequence identity with other badnavirus sequences (Supplementary Table S2). Phylogenetic trees based on full genome sequences and on the RNase region were generated by the maximum-likelihood method, using MEGA version 7.0, and the robustness of each internal branch was estimated by 1000 bootstrap replicates. Both the full-genome phylogenetic tree (Fig. 1B) and the tree based on RNase nucleotide sequences (Fig. 1C) show that JuBWS clusters with JuMaV. The data suggest that JuBWS should be considered a member of a new species in the genus Badnavirus.

(A) Schematic diagram of the JuBWS genome structure (right panel). The tRNAMet binding site was set as the start site of the JuBWS genome. ORFs (ORF1, ORF2, and ORF3) are the three putative open reading frames of the JuBWS genome (left panel). tRM, tRNAMet binding site; DUF1319, domain of unknown function; ZnF, zinc-finger domain; AP, pepsin-like aspartate protease domain; RT, reverse transcriptases (RTs) from retrotransposons and retroviruses domain; RNase H, ribonuclease-H-like superfamily domain. (B) Phylogenetic tree constructed based on complete genome nucleotide sequences. (C) Phylogenetic tree based on nucleotide sequences of the RT+RNase H region. The viruses that were used for analysis are as follows: GBV 1, grapevine badnavirus 1; FBV 1, fig badnavirus 1; CiYMV, citrus yellow mosaic virus; HiBV, hibiscus bacilliform virus; DBALV, dioscorea bacilliform AL virus; CMMV, cacao mild mosaic virus; CSSGQV, cacao swollen shoot Ghana Q virus; BSCAV, banana streak CA virus; SCBGAV, sugarcane bacilliform Guadeloupe A virus; JuMaV, jujube mosaic-associated virus; BVF, blackberry virus F; PVBV, pelargonium vein banding virus; DrMV, dracaena mottle virus; LBBV, lucky bamboo bacilliform virus; ChMV, chestnut mosaic virus; BLRaV, birch leaf roll-associated virus; EpMoaV, epiphyllum mottle-associated virus; BCVBV, Bougainvillea spectabilis chlorotic vein-banding virus; BCVBV, Bougainvillea spectabilis chlorotic vein-banding virus from Taiwan, complete genome. The corresponding DNA sequence from sweet potato caulimo-like virus (SPCV) was used as an outgroup in each phylogenetic tree
The full genome of JuBWS, with a length of 6450 bp, is shorter than those of all other previously reported badnaviruses, and the JuBWS genome has one less open reading frame (ORF3a) than JuMaV.
JuBWS was detected in both leaf and fruit samples (Supplementary Fig. S3). The establishment of a high-efficiency detection method for JuBWS will help to monitor the geographical distribution of this virus in Xinjiang and other provinces in China where jujube is cultivated. This method will also be useful for investigating variations between different field isolates and identification of JuBWS-resistant jujube varieties. In future research, we will explore the prevention of viral transmission by vectors and the resistance of plants to these viruses.
References
Liu MJ, Liu P, Liu GN (2013) Advances of research on germplasm resources of Chinese jujube. Acta Hortic. https://doi.org/10.17660/ActaHortic.2013.993.1
Du K, Liu S, Chen Z et al (2017) Full genome sequence of jujube mosaic-associated virus, a new member of the family Caulimoviridae. Arch Virol 162:3221–3224. https://doi.org/10.1007/s00705-017-3438-6
Medberry SL, Lockhart BEL, Olszewski NE (1990) Properties of Commelina yellow mottle virus’s complete DNA sequence, genomic discontinuities and transcript suggest that it is a pararetrovirus. Nucl Acids Res 18:5505–5513. https://doi.org/10.1093/nar/18.18.5505
Borah BK, Sharma S, Kant R et al (2013) Bacilliform DNA-containing plant viruses in the tropics: commonalities within a genetically diverse group: tropical badnaviruses and tungroviruses. Mol Plant Pathol 14:759–771. https://doi.org/10.1111/mpp.12046
Chabannes M, Gabriel M, Aksa A et al (2021) Badnaviruses and banana genomes: a long association sheds light on Musa phylogeny and origin. Mol Plant Pathol 22:216–230. https://doi.org/10.1111/mpp.13019
Chambers GA, Donovan NJ, Bodaghi S et al (2018) A novel citrus viroid found in Australia, tentatively named citrus viroid VII. Arch Virol 163:215–218. https://doi.org/10.1007/s00705-017-3591-y
Muller E, Sackey S (2005) Molecular variability analysis of five new complete cacao swollen shoot virus genomic sequences. Arch Virol 150:53–66. https://doi.org/10.1007/s00705-004-0394-8
Tahir MN, Bolus S, Grinstead SC et al (2021) A new virus of the family Tombusviridae infecting sugarcane. Arch Virol 166:961–965. https://doi.org/10.1007/s00705-020-04908-9
Kazmi SA, Yang Z, Hong N et al (2015) Characterization by small RNA sequencing of taro bacilliform CH Virus (TaBCHV), a novel badnavirus. PLoS ONE 10:e0134147. https://doi.org/10.1371/journal.pone.0134147
Marais A, Umber M, Filloux D et al (2020) Yam asymptomatic virus 1, a novel virus infecting yams (Dioscorea spp.) with significant prevalence in a germplasm collection. Arch Virol 165:2653–2657. https://doi.org/10.1007/s00705-020-04787-0
Diaz-Lara A, Mosier NJ, Stevens K et al (2020) Evidence of rubus yellow net virus integration into the red raspberry genome. Cytogenet Genome Res 160:329–334. https://doi.org/10.1159/000509845
Laney AG, Hassan M, Tzanetakis IE (2012) An integrated badnavirus is prevalent in fig germplasm. Phytopathology® 102:1182–1189. https://doi.org/10.1094/PHYTO-12-11-0351
Xu D, Mock R, Kinard G, Li R (2011) Molecular analysis of the complete genomic sequences of four isolates of Gooseberry vein banding associated virus. Virus Genes 43:130–137. https://doi.org/10.1007/s11262-011-0614-8
Teycheney P-Y, Geering ADW, Dasgupta I et al (2020) ICTV virus taxonomy profile: caulimoviridae. J Gen Virol 101:1025–1026. https://doi.org/10.1099/jgv.0.001497
Kidanemariam DB, Sukal AC, Abraham AD et al (2018) Identification and molecular characterization of Taro bacilliform virus and Taro bacilliform CH virus from East Africa. Plant Pathol 67:1977–1986. https://doi.org/10.1111/ppa.12921
Bhat A, Hohn T, Selvarajan R (2016) Badnaviruses: the current global scenario. Viruses 8:177. https://doi.org/10.3390/v8060177
Zerbino DR, Birney E (2008) Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. https://doi.org/10.1101/gr.074492.107
Peng Y, Leung HCM, Yiu SM, Chin FYL (2012) IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28:1420–1428. https://doi.org/10.1093/bioinformatics/bts174
Bateman A, Coggill P, Finn RD (2010) DUFs: families in search of function. Acta Crystallogr F Struct Biol Cryst Commun 66:1148–1152. https://doi.org/10.1107/S1744309110001685
Acknowledgements
Financial support was provided by the Research and Demonstration of Key Technologies for Blockade Prevention and Control of Jujube Virus Diseases in Xinjiang (grant number: 2018E02026) and “Excellent Postdoctoral Professionals and General Funding Staff of Living Subsidies” from the Department of Human Resources and Social Security of Xinjiang Uygur Autonomous Region.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: Elvira Fiallo-Olivé.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liu, B., Zhang, G., Song, D. et al. Complete genome sequence of a novel virus belonging to the genus Badnavirus in jujube (Ziziphus jujuba Mill.) in China. Arch Virol 167, 1885–1888 (2022). https://doi.org/10.1007/s00705-022-05482-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-022-05482-y