
Abstract
As essential transfer carriers for cell-to-cell communication and genetic material, exosomes carry microRNAs that participate in the regulation of various biological processes. MicroRNAs are a type of single-stranded noncoding RNA that bind to specific target gene mRNAs to degrade or inhibit their translation, thereby regulating target gene expression. Although it is known that a variety of microRNAs are involved in the viral infection process, there are few reports on specific microRNAs involved in porcine epidemic diarrhea virus (PEDV) infection. In this study, we isolated and identified exosomes in PEDV-infected Vero E6 cells. Using transcriptomics technology, we found that miRNA-328-3p was significantly downregulated in exosomes following PEDV infection. Moreover, exosomal miRNA-328-3p inhibited infection by PEDV by targeting and inhibiting tight junction protein 3 (TJP-3/ZO-3) in recipient cells. Our findings provide evidence that, after infecting cells, PEDV downregulates expression of miRNA-328-3p, and the resulting reduced inhibition of the target protein ZO-3 helps to enhance PEDV infection. These results provide new insight for understanding the regulatory mechanism of PEDV infection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Porcine epidemic diarrhea virus (PEDV), which belongs to the genus Alphacoronavirus, family Coronaviridae, is an enveloped, single-stranded, positive-sense RNA virus. Porcine epidemic diarrhea (PED) is an acute and highly contagious enteric disease caused by PEDV that is characterized by watery diarrhea, vomiting, and high mortality in suckling and nursing pigs. Some studies have shown that the S1 protein of PEDV is one of the most critical functional proteins contributing to apoptosis [1]. The PEDV envelope (E) protein, membrane (M) protein, nucleocapsid (N) protein, open reading frame 3 (ORF3) protein, nonstructural protein 1 (NSP1), NSP3, NSP7, NSP14, NSP15, and NSP16 suppress interferon (IFN) activity to promote replication [2,3,4,5]. However, Luo et al. demonstrated that occludin plays an essential role in PEDV infection. Moreover, PEDV entry and occludin internalization appear to be closely coupled [6]. Occludin is a critical tight junction (TJ) protein, and TJs are essential for the maintenance and repair of the intestinal mucosal barrier. Members of the zonula occludens (ZO) family (including ZO-1–3) and the occludin and claudin families (including claudin-1–24 in pig) are essential TJ proteins. Zong et al. found that occludin and claudin-4 play an essential role in PEDV infection in piglets [7]. Structurally impaired TJ and adhesion junction (AJ) proteins are involved in the pathogenesis of PEDV [8]. Moreover, coinfection with PEDV and transmissible gastroenteritis virus (TGEV) results in more damage to tight corners and remodelling of microfilaments than infection with PEDV or TGEV alone [9]. In essence, the villous epithelium of the small intestine can maintain the integrity of the intestinal wall through tight junction proteins, and PEDV infection has been found to destroy epithelial cells of intestinal villi, reduce intestinal absorption capacity, and cause malabsorptive diarrhea.
Exosomes are small vesicles derived from different types of cells that are actively released into the extracellular space. Exosomes transport proteins, mRNA, miRNA, and nucleic acids between cells and serve as mediators of intercellular communication [10]. Accumulating evidence suggests that exosomes derived from cells infected by certain viruses selectively mediate cell-to-cell communication and virus transmission [11, 12], altering the microenvironment at the site of viral infection, transmitting cell growth and transformation signals, and participating in various pathological processes in target cells [13, 14]. Hence, it may be argued that exosomes play an essential role in virus infection and pathogenicity. In this study, exosomes were isolated and purified from cells infected with PEDV and from mock-infected (negative control) cells, and transcriptomics sequencing was applied to explore the role of exosomes in the pathogenicity and virulence of PEDV.
Materials and methods
Cells and virus
Vero E6 cells were provided by the Military Veterinary Research Institute (Changchun, China). The classical PEDV CV777 strain was purchased from Collection of Chinese Bacteria and Virus Species (Beijing, China). In all PEDV culture experiments, 0.05% trypsin was used.
Isolation, purification, and characterization of exosomes
Vero E6 cells were cultured in DMEM (HyClone, USA) containing 10% foetal bovine serum until the cells grew to 90% confluence. The culture medium was then discarded, and the cells were washed with phosphate-buffered saline (PBS). The experimental group was infected with the standard PEDV vaccine strain CV777 at a multiplicity of infection (MOI) of 1. In the mock infection group, the same volume of DMEM was added. After incubation for 1 hour at 37 °C, serum-free DMEM was added, incubation was continued for another 48 hours. The supernatant was collected and stored at −80 °C. We used exosome isolation kits (Umibio Co., Ltd. Shanghai, China) for exosome isolation. Briefly, the sample, which had been stored at -80 °C, was centrifuged at 10,000 × g for 10 min at 4 °C to remove cell debris, and the supernatant was filtered through a 0.22-μm filter. A 30-kDa ultrafiltration tube was used to concentrate the exosomes. In accordance with the manufacturer’s instructions, a half volume of exosome precipitation reagent was added to the sample, which was incubated overnight at 4 °C and centrifuged at 4 °C and 10,000 × g for 1 h. The sediment at the bottom of the centrifuge tube was resuspended in PBS, transferred to the exosome purification filter (EPF) column, and centrifuged at 4,000 × g for 10 min at 4 °C to collect the liquid at the bottom of the tube. The product was further purified by ultracentrifugation [15]. Finally, the exosomes were characterized by transmission electron microscopy (TEM) and western blotting.
RNA extraction and transcriptome sequencing
In accordance with the manufacturer’s instructions, total RNA was extracted from exosomes obtained from different treatment groups using a Simply P Total RNA Extraction Kit (BioFlux, China). The quality of the RNA preparation was assessed using an Agilent Small RNA Kit with an Agilent 2100 Bioanalyzer. Small RNAs were used for library construction using an Ion Total RNA-Seq Kit v2 (Life Technologies Corp.). Bioanalysis was performed using an Agilent 2100 Eukaryote Total RNA Pico Series II, and RNA-Seq analysis was carried out using an Illumina HiSeq 2500 Sequencing System.
Quantitative real-time RT-PCR analysis
Total RNA from exosomes or cells was extracted and purified using a BioFlux RNA Extraction Kit (BioFlux Company, Tokyo, Japan). For analysis of exosomal microRNA (miR-103a, let-7d, miR-411, miR-345, miR-328-3P) expression, 0.5 mg of total RNA was reverse transcribed using a PrimeScript® 1st Strand cDNA Synthesis Kit (BioFlux Company, Tokyo, Japan) according to the standard protocol. For mRNA (PEDV) expression analysis, complementary DNA (cDNA) was synthesized using a PrimeScript® 1st Strand cDNA Synthesis Kit (BioFlux Company, Tokyo, Japan) according to the manufacturer’s protocol. RT-qPCR was performed using SYBR® Premix Ex TaqTM II (TaKaRa, Dalian, China) according to the manufacturer’s instructions. The sequences of the primers used are shown in Table 1. Briefly, 2.0 μL of cDNA template was added to a total volume of 20.0 μL containing 10.0 μL of SYBR®Premix Ex TaqTM II (TaKaRa, Dalian, China), 0.4 μL of ROX reference dye, 6.0 μL of ddH2O, and 0.8 μL of the forward and reverse primers. The thermal cycling conditions were as follows: (1) predenaturation (30 s at 95 ℃) and (2) amplification and quantification (40 cycles of 30 s at 94 ℃ and 30 s at 60 ℃). Relative expression was assessed as the ratio of expression of the target gene to that of a housekeeping gene used as an internal standard, using the formula 2−△△t. Relative expression was normalized and is given as a ratio relative to the control group.
Western blot analysis
Exosomal proteins and PEDV nucleocapsid protein (PEDV-N) were detected by western blotting. Briefly, protein concentrations of exosomes and PEDV-N were determined using a BCA Protein Detection Kit (Biouniquer Technology Co., LTD, Nanjing, China), and 50 μg of protein lysate from each sample was used for further analysis. After SDS-PAGE electrophoresis, proteins were electrotransferred to a polyvinylidene difluoride (PVDF) membrane. Next, 5% skimmed milk powder was used to block nonspecific antibody binding to the membrane for 1 h. Rabbit anti-CD63/TSG101 monoclonal antibody (Abcam, UK), rabbit anti-ZO3 monoclonal antibody (ImmunoWay, China), mouse anti-PEDV-N monoclonal antibody (Medgene Labs, UK), and β-actin primary antibodies (ImmunoWay, China) were added, and the membrane was kept at 4 ℃ overnight. The membranes were washed three times with TBST, and HRP-labelled goat anti-rabbit IgG and goat anti-mouse IgG (Proteintech, Chicago, USA) were added. The membranes were incubated at room temperature for 1 h and then washed three times with TBST. Finally, the blots were visualized using an ECL kit (Beyotime, Shanghai, China); signal intensity was quantified using greyscale analysis software (Image Tool 3.00).
Virus titre determination and cytopathic observation
Each sample was serially diluted tenfold, and 100 μL of each dilution was added per well to each of eight columns of a microtiter plate, followed by the addition of 100 μL of DMEM with 2% FBS. A control of virus-free medium was included, and the plate was kept at 37 °C in 5% CO2. The virus median tissue culture infectious dose (TCID50) was calculated using the Reed–Muench formula [??REFERENCE??]. Exosomes were extracted from PEDV-CV777-infected Vero cells, and PEDV-infected Vero cells with or without added exosomes were incubated in 1.3% methylcellulose DMEM for 48 h. Then, 4% formaldehyde was added overnight at room temperature, and the cells were stained with crystal violet staining solution. Cytopathic effect (CPE) was observed using a Leica inverted fluorescence microscope DMi8 (Leica, Germany).
Plasmid construction
The GP-miRGLO vector was used for the construction of ZO-3-WT/ZO-3-MUT/PC. First, we used the primers listed in Table 1 to clone the target gene into the cloning site of the GP-miRGLO vector. The resulting plasmid was introduced by transformation into competent cells and then extracted using a Plasmid Large-Scale Extraction Kit (Sigma-Aldrich, China) and used for subsequent experiments. The primers shown in Table 1 were used to amplify the complete sequence of the ZO-3 gene from Vero E6 cells for cloning into the pLVX-IRES-ZsGreen1 vector (Clontech, USA). To ensure the fidelity of the gene sequence, we followed the appropriate operating steps for the production and packaging of lentiviral vectors [16]. Vero E6 cells were transfected with the lentiviral vector at 80% confluence. After 48 h, the cells and supernatant were harvested for experiments.
Cell transfection
Lipofectamine 2000 (Invitrogen) was used for transfection when cells had grown to 80% confluence in a 24-well plate. Briefly, DMEM with 10% FBS was added to a 6-well plate, and when the cell coverage reached 60%, the medium was replaced with DMEM. miRNA, inhibitor (50 nM), ZO-3-WT/ZO-3-MUT/PC (2 μg), and Lipofectamine 2000 were thoroughly mixed with an equal volume of DMEM (500 µl) in a centrifuge tube, and the mixture was incubated at 37 °C and 5% CO2 for 24 h. A dual-luciferase experiment was then performed. After transfection for 24 h with miRNA mimics and inhibitors (50 nM), PEDV was added at an MOI of 1. The siRNA (100) shown in Table 1 was introduced into cells using a lentiviral vector, and after 24 hours, the cells were infected with PEDV at an MOI of 1 and subsequent experiments were carried out.
Dual-luciferase reporter gene activity assay
A Dual-Luciferase Reporter Assay System Kit (Promega, USA) was used to detect luciferase activity in accordance with the manufacturer’s protocol. Vero E6 cells were seeded into 24-well plates 1 day prior to transfection with 1.6 µg of the GP-miRGLO reporter construct containing ZO-3-WT/ZO-3-MUT/PC (the primer sequences are shown in Table 1) together with 10 µL of miR-328-3p mimic/mock, using Lipofectamine 2000 (Thermo Fisher Scientific, USA). The cells were washed and lysed with passive lysis buffer from the Dual-Luciferase Reporter Assay System (Promega Corp, USA) at 48 h after transfection. Luciferase activity in each cell lysate was measured using a Multifunctional Microplate Reader (Tecan, USA). Relative luciferase activity was first normalized to Renilla luciferase activity and then compared with that of the respective control.
Results
Exosomes isolated from PEDV-infected cells enhance PEDV virulence
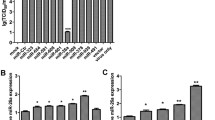
To study the role of exosomes in PEDV-infected cells, we first isolated exosomes from the supernatants of infected cells. Using TEM, exosomes were observed to have a typical “cup-shaped” morphology and a crescent-shaped outer membrane (Fig. 1A). We detected two exosomal marker proteins, CD63 and TSG101, in the PEDV infection group and in the mock-infected group. Expression of CD63 increased significantly after PEDV infection (Fig. 1B), and we found that adding PEDV-infected exosomes after PEDV infection promoted virus replication (Fig. 1C). Moreover, the level of PEDV mRNA expression gradually increased with the addition of exosomes (Fig. 1D). Cytopathic changes were revealed by crystal violet staining after the addition of exosomes, and the cells were infected with PEDV (Fig. 1E), showing that exosomes promoted PEDV infection.

Isolation of exosomes and their effect on PEDV virulence. (A) Image of exosomes observed by TEM after PEDV infection. (B) Expression of the marker proteins CD63 and TSG101 in exosomes of the PEDV infection group and the mock-infected group detected by western blotting. (C) The effect of added exosomes on expression of PEDV RNA by RT-qPCR. (D) The effect of different volumes of added exosomes on expression of PEDV RNA, as shown by RT-qPCR. (E) Effects of exosomes on CPE after cells were infected with PEDV, as shown by crystal violet staining. All experiments were repeated three times. Error bars represent the mean with SD. ns < 0.1234; *, p < 0.0332; **, p < 0.0021
PEDV infection affects miRNA levels in exosomes
Exosome extracts were subjected to microRNA transcriptome sequencing, which revealed 861 differentially expressed microRNAs (Fig. 2A). When selecting those with a p-value range of 0.001-0.01 for statistical analysis, we detected 26 differentially expressed microRNAs (Fig. 2B). We then used the enrichment factor, the Q value, and the number of genes annotated to pathways to assess the degree of pathway enrichment (Fig. 2C). According to summary statistical analysis of the results, we found tight junctions (TJs) to be of great significance. We screened microRNAs targeting the TJ pathway from microRNAs with significant differential expression and found eight differentially expressed genes related to the TJ pathway, with miRNA-328-3p being significantly downregulated (Fig. 2D). We randomly selected five differentially expressed microRNAs and verified their expression by RT-qPCR. The results were consistent with the sequencing results, and miRNA-328-3p significantly downregulated PEDV mRNA expression levels (Fig. 2E). These results indicate that the TJ pathway may play an important role in PEDV infection and that miRNA-328-3p may play an essential regulatory role.

Expression of miRNA-328-3p by transcriptome sequencing and its involvement in the TJ pathway. (A) We used the log10 (TPM+1) value to perform hierarchical cluster analysis of differentially expressed microRNAs with the same or similar expression behaviour. We then employed online analysis software (http://www.metaboanalyst.ca) to produce a heat map of differentially expressed microRNA clusters. (B) To further study the relationship between PEDV infection and exosomal microRNA expression, we screened for microRNAs with significant differences in expression, using a p-value of 0.001-0.01 as an indicator of significance. (C) The degree of pathway enrichment was measured using the enrichment factor, Q value, and number of genes annotated to pathways. We first analysed KEGG results using the KOBAS web tool and then used ggplot2 for drawing. We finally obtained a scatter plot of target gene KEGG pathway enrichment. (D) Eight microRNAs with significant differential expression were screened from among the microRNAs targeting the TJ pathway. All experiments were repeated three times. (E) We randomly selected five microRNAs for RT-qPCR verification of the results. ***, p < 0.0002. The data were derived from three independent experiments, each performed in triplicate.
The effect of miRNA-328-3p on PEDV infection
To determine whether Vero cells were successfully transfected with miR-328-3p, we used miR-328-3p mimic/inhibitor labelled with fluorescein amide (FAM) for transfection. Fluorescence microscopy analysis showed that the transfection was successful based on observation of a green fluorescent signal in the cytoplasm that was not observed in mock-transfected cells (Fig. 3A). Compared with the control, addition of the miR-328-3p inhibitor resulted in a significant increase in the expression level of PEDV N mRNA and protein. In contrast, addition of miR-328-3p mimic substantially reduced the expression level of the PEDV N protein (Fig. 3B, C, and D). Furthermore, addition of a miR-328-3p inhibitor substantially increased the viral titre of PEDV. Compared with the control, addition of miR-328-3p mimic did not significantly affect the viral titre (Fig. 3E).

Inhibition of PEDV infection by miRNA-328-3p. (A) Vero cells were transfected with miR-328-3p mimic/inhibitor with a 5`FAM fluorophore. (B) The expression level of the PEDV N protein was estimated by western blotting. (C) We used ImageJ software to normalize PEDV N protein levels according to β-actin protein levels. (D) The RNA expression level of PEDV was determined by RT-qPCR. (E) The effect of miR-328-3p mimic/inhibitor on PEDV virus titre after transfection. ns < 0.1234; *, p < 0.0332; **, p < 0.0021; ****, p < 0.0001. The data were derived from three independent experiments, each performed in triplicate.
miR-328-3p can target the ZO-3 protein in the TJ pathway
To confirm that ZO-3 is a direct downstream target of miR-328-3p, we first performed western blotting and RT-qPCR analysis using extracts from Vero cells that were transfected with miR-328-3p mimic/inhibitor or mock transfected. The results showed that the miR-328-3p mimic significantly reduced mRNA and protein expression levels of ZO-3 compared with the mock-transfected control but that the miR-328-3p-inhibitor significantly increased levels of ZO-3 (Fig. 4A-C). These results indicate that miR-328-3p has a negative regulatory effect on the ZO-3 protein with respect to the TJ pathway. At the same time, we verified the regulatory relationship between miR-328-3p and ZO-3 using the dual-luciferase reporter gene system. Cells with wild-type ZO-3 that were transfected with miR-328-3p and the positive control (PC) showed significantly decreased luciferase activity, but miR-328-3p had no effect on mutant ZO-3 (Fig. 4D). These results demonstrate that the ZO-3 protein is regulated directly by miR-328-3p.

The regulatory effect of exosomal miR-328-3p on the ZO-3 protein. (A) Expression of ZO-3 protein detected by western blotting. (B) Normalization of the ZO-3 protein with endogenous β-actin using ImageJ software. (C) RT-qPCR detection of the genomic RNA expression level of ZO-3. (D) The relationship between miR-328-3p and ZO-3 detected using a dual-luciferase reporter gene activity assay. ns < 0.1234; ***, p < 0.0002. The data are representative of at least three independent experiments, each performed in triplicate.
Promotion of PEDV infection by upregulation of the ZO-3 protein
We demonstrated that miR-328-3p can inhibit infection of Vero cells by PEDV. To determine whether the regulation of PEDV infection by miR-328-3p requires the ZO-3 protein, we silenced or overexpressed ZO-3 in Vero cells and detected PEDV nucleocapsid protein (PEDV N) and mRNA and measured the virus titre. The results showed that expression of the ZO-3 protein was significantly lower in cells treated with an siRNA than in the control. Simultaneously, expression of the viral N protein and viral mRNA was significantly lower than in the control group, indicating that a decrease in ZO-3 protein expression has a negative effect on viral N protein and RNA expression (Fig. 5A-C). Moreover, a similar negative effect on the virus titre was observed (Fig. 5D). In contrast, overexpression of the ZO-3 protein in virus-infected cells resulted in a significant increase in the expression levels of the viral N protein and viral mRNA in infected cells (Fig. 5E-G), and the virus titre increased significantly (Fig. 5H). These results provide strong evidence that the ZO-3 protein facilitates PEDV infection.

Involvement of ZO-3 in PEDV infection. (A, E) Vero cells were transfected with Si-ZO-3, control interfering RNA (si-Ctrl), over-ZO-3, or control overexpression vector (over-Ctrl). At 48 h postinfection, cell lysates were prepared to measure ZO-3 and PEDV N protein expression. (B, F) Normalization of ZO-3 and PEDV N protein expression to that of endogenous β-actin, carried out using ImageJ software. (C, G) PEDV genomic RNA levels in si-ZO-3- and ZO-3-transfected Vero cells, analysed by RT-qPCR. (D, H) Viral titres in infected cells, determined at 48 h postinfection. *, p < 0.0332, **; p < 0.0021; ***, p < 0.0002. Data are representative of at least three independent experiments, each performed in triplicate.
Discussion
In previous studies, it was shown that exosomes carry various intracellular regulatory RNAs, including miRNA, sncRNA, and siRNA [17]. Furthermore, the components of exosomes, which may be proteins or noncoding RNAs, change after virus infection [18]. Exosomes were initially regarded as waste products of cells before they received extensive attention from the scientific community. Studies in recent years have found that they represent a novel means of cell-to-cell communication. The identification of exosomes is usually based on electron microscopy and western blotting to detect exosomal marker proteins (such as CD63, CD81, TSG101, annexin 5, and ICAM1) [19]. We found that PEDV infection increased expression of CD63. PEDV infection may promote cells to secrete exosomes, but the concentration of exosomes still needs to be determined. When we combined the same volume of exosomes from PEDV-infected and uninfected cells, we found that adding PEDV-infected cell exosomes upregulated expression of PEDV genes and enhanced the CPE caused by PEDV in these cells. Hence, there appear to be certain exosomes that promote PEDV infection. Overall, the composition of exosomes is complex [20], but we did not detect infectious virus particles in exosomes from PEDV-infected cells. However, it still needs to be investigated whether exosomes contain PEDV gene fragments or viral proteins.
We performed transcriptome sequencing on extracted exosomes and found 862 microRNAs that were differentially expressed. The TJ pathway may play an essential role in the PEDV infection process according to KEGG pathway annotation, and miRNA-328-3p is related to the TJ pathway. MicroRNAs can affect interactions between viruses and host cells in a variety of ways. In previous studies, it was shown that miRNA-221-5p can target the PEDV genome after infection and activate the NF-κB pathway to inhibit viral replication [21]. Moreover, some microRNAs can regulate related proteins in the interferon pathway, thereby affecting interferon expression to be conducive to virus proliferation [22]. In a study on the relationship between miR-122 and hepatitis C virus (HCV), researchers found that miR-122 can promote HCV replication [23]. Our research showed that miRNA-328-3p downregulates PEDV mRNA and the N protein and reduces the PEDV titre. Thus, miRNA-328-3p exerts an inhibitory effect on PEDV.
Claudin is widely present in various epithelial cells. It is mainly responsible for sealing the intercellular space to prevent the random entry and exit of the epithelial layer to maintain integrity of the epithelial barrier [8]. It has been reported that occludin is essential for PEDV infection [6]. Expression of occludin is also related to the sensitivity of PEDV to cells [6]. Although PEDV and occludin affect each other, there is no direct interaction between them [6]. PEDV inhibits expression of ZO-1 in the early stage of infection [9], but there is no research showing that ZO-3 is related to PEDV infection. In our study, addition of miRNA-328-3p significantly inhibited the expression of ZO-3, with ZO-3 apparently playing a significant role in PEDV infection. When ZO-3 was inhibited, PEDV mRNA and N protein expression levels were significantly reduced. This may be because ZO-3 is involved in the process of virus internalization [24]. In addition, overexpression of the ZO-3 protein significantly increased the PEDV titre. Interestingly, occludin overexpression also increases the sensitivity of cells to PEDV [6]. In general, ZO-3 has a positive regulatory effect on PEDV infection.
Based on these results, we conclude that PEDV infection downregulates expression of miRNA-238-3p in exosomes secreted by cells. Such downregulation of miRNA-238-3p upregulates ZO-3 protein expression and increases the infectivity of PEDV. The mechanism by which PEDV regulates expression of miRNA-238-3p and the interaction between the ZO-3 protein and PEDV in cells is not fully understood, and the interactions between PEDV and the host appear to be complex. Therefore, further work is required [25]. In short, our study provides insight into the infection and pathogenicity of PEDV and contributes to research on the mechanism by which PEDV damages the intestine.
References
Chen Y, Zhang Z, Li J et al (2018) Porcine epidemic diarrhea virus S1 protein is the critical inducer of apoptosis. Virol J 15:170. https://doi.org/10.1186/s12985-018-1078-4
Wu Y, Zhang H, Shi Z et al (2020) Porcine epidemic diarrhea virus nsp15 antagonizes interferon signaling by RNA degradation of TBK1 and IRF3. Viruses 12:599. https://doi.org/10.3390/v12060599
Kaewborisuth C, Koonpaew S, Srisutthisamphan K et al (2020) PEDV ORF3 independently regulates IκB kinase β-mediated NF-κB and IFN-β promoter activities. Pathogens 9:376. https://doi.org/10.3390/pathogens9050376
Zhang Q, Shi K, Yoo D (2016) Suppression of type I interferon production by porcine epidemic diarrhea virus and degradation of CREB-binding protein by nsp1. Virology 489:252–268. https://doi.org/10.1016/j.virol.2015.12.010
Zhang Q, Ke H, Blikslager A et al (2018) Type III interferon restriction by porcine epidemic diarrhea virus and the role of viral protein nsp1 in IRF1 signaling. J Virol. https://doi.org/10.1128/JVI.01677-17
Luo X, Guo L, Zhang J et al (2017) Tight junction protein occludin is a porcine epidemic diarrhea virus entry factor. J Virol. https://doi.org/10.1128/JVI.00202-17
Zong QF, Huang YJ, Wu LS et al (2019) Effects of porcine epidemic diarrhea virus infection on tight junction protein gene expression and morphology of the intestinal mucosa in pigs. Pol J Vet Sci 22:345–353. https://doi.org/10.24425/pjvs.2019.129226
(2015) Structural alteration of tight and adherens junctions in villous and crypt epithelium of the small and large intestine of conventional nursing piglets infected with porcine epidemic diarrhea virus. Vet Microbiol 177:373–378. https://doi.org/10.1016/j.vetmic.2015.03.022
Zhao S, Gao J, Zhu L, Yang Q (2014) Transmissible gastroenteritis virus and porcine epidemic diarrhoea virus infection induces dramatic changes in the tight junctions and microfilaments of polarized IPEC-J2 cells. Virus Res 192:34–45. https://doi.org/10.1016/j.virusres.2014.08.014
Valadi H, Ekström K, Bossios A et al (2007) Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9:654–659. https://doi.org/10.1038/ncb1596
Chivero ET, Stapleton JT (2015) Tropism of human pegivirus (formerly known as GB virus C/hepatitis G virus) and host immunomodulation: insights into a highly successful viral infection. J Gen Virol 96:1521. https://doi.org/10.1099/vir.0.000086
Wang T, Fang L, Zhao F et al (2018) Exosomes mediate intercellular transmission of porcine reproductive and respiratory syndrome virus. J Virol. https://doi.org/10.1128/JVI.01734-17
Bernard MA, Zhao H, Yue SC et al (2014) Novel HIV-1 MiRNAs stimulate TNFα release in human macrophages via TLR8 signaling pathway. PLoS ONE 9:e106006. https://doi.org/10.1371/journal.pone.0106006
Dreux M, Garaigorta U, Boyd B et al (2012) Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 12:558–570. https://doi.org/10.1016/j.chom.2012.08.010
Zhang K, Xu S, Shi X et al (2019) Exosomes-mediated transmission of foot-and-mouth disease virus in vivo and in vitro. Vet Microbiol 233:164–173. https://doi.org/10.1016/j.vetmic.2019.04.030
Li R, Guo L, Gu W et al (2016) Production of porcine TNFα by ADAM17-mediated cleavage negatively regulates porcine reproductive and respiratory syndrome virus infection. Immunol Res 64:711–720. https://doi.org/10.1007/s12026-015-8772-8
Vojtech L, Woo S, Hughes S et al (2014) Exosomes in human semen carry a distinctive repertoire of small non-coding RNAs with potential regulatory functions. Nucleic Acids Res 42:7290–7304. https://doi.org/10.1093/nar/gku347
Chahar HS, Bao X, Casola A (2015) Exosomes and their role in the life cycle and pathogenesis of RNA viruses. Viruses 7:3204–3225. https://doi.org/10.3390/v7062770
Mathivanan S, Ji H, Simpson RJ (2010) Exosomes: extracellular organelles important in intercellular communication. J Proteomics 73:1907–1920. https://doi.org/10.1016/j.jprot.2010.06.006
Zheng B, Zhou J, Wang H (2020) Host microRNAs and exosomes that modulate influenza virus infection. Virus Res 279:197885. https://doi.org/10.1016/j.virusres.2020.197885
Zheng H, Xu L, Liu Y et al (2018) MicroRNA-221-5p inhibits porcine epidemic diarrhea virus replication by targeting genomic viral RNA and activating the NF-κB pathway. Int J Mol Sci 19:3381. https://doi.org/10.3390/ijms19113381
Pedersen IM, Cheng G, Wieland S et al (2007) Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 449:919–922. https://doi.org/10.1038/nature06205
Henke JI, Goergen D, Zheng J et al (2008) microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J 27:3300–3310. https://doi.org/10.1038/emboj.2008.244
Stamatovic SM, Johnson AM, Sladojevic N et al (2017) Endocytosis of tight junction proteins and the regulation of degradation and recycling. Ann N Y Acad Sci 1397:54–65. https://doi.org/10.1111/nyas.13346
González-Mariscal L, Tapia R, Chamorro D (2008) Crosstalk of tight junction components with signaling pathways. Biochim Biophys Acta 1778:729–756. https://doi.org/10.1016/j.bbamem.2007.08.018
Acknowledgements
The present work was supported by the National Natural Science Foundation of China (32072857, China).
Author information
Authors and Affiliations
Contributions
HZ, JY, KW, and GH conceived and designed the experiments. HZ, JY, and WL performed the experiments. HZ and JY analyzed the data. ZC, DL, JN, SZ, YZ, YG, and QZ contributed reagents/materials/analysis tools. HZ, and JY wrote the paper. GH, WL, and KW requested financial support. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Handling Editor: Sheela Ramamoorthy.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhao, H., Yang, J., Wang, Q. et al. Exosomal miRNA-328-3p targets ZO-3 and inhibits porcine epidemic diarrhea virus proliferation. Arch Virol 167, 901–910 (2022). https://doi.org/10.1007/s00705-022-05364-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-022-05364-3