
Abstract
Porcine reproductive and respiratory syndrome (PRRS) is a highly contagious infectious disease caused by porcine reproductive and respiratory syndrome virus (PRRSV), which inflicts major economic losses on the global pig farming industry. Based on its similarity to highly pathogenic strains, the GDzj strain isolated in this study was predicted to be highly pathogenic. We therefore analyzed the pathogenicity of this strain experimentally in piglets. All piglets challenged with this virus experienced fever or high fever, loss of appetite, decreased food intake, daily weight loss, shortness of breath, and listlessness, and the necropsy results showed that they had experienced severe interstitial pneumonia. We then used the BAC system to construct a full-length cDNA infectious clone of GDzj, and the rescued virus displayed in vitro proliferation characteristics similar to those of the parental PRRSV strain. In summary, we successfully isolated a highly pathogenic PRRSV strain and constructed a full-length infectious cDNA clone from it, thereby providing an effective reverse genetics platform for further study of viral pathogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Porcine reproductive and respiratory syndrome (PRRS) is a highly contagious infectious disease caused by porcine reproductive and respiratory syndrome virus (PRRSV), a member of the family Arteriviridae [4]. It is an enveloped, positive-sense, single-stranded RNA virus. The genome of this virus is approximately 15 kb long, with a methylated ‘cap’ structure at the 5' end, a polyadenylic acid (poly A) tail at the 3' end, and at least 10 open reading frames (ORFs) [26, 30]. Most of the adjacent ORF gene structures in PRRSV overlap. ORF1, which accounts for 80% of the total genome length, encodes proteins involved in viral replication. This largest segment of the genome includes ORF1a and ORF1b, which encode two polyprotein types, pp1a and pp1ab. The pp1a polyprotein is processed to form nine non-structural proteins (NSPs)–NSP1α, NSP1β, and NSP2–NSP8–whereas pp1ab is processed to form NSP9–NSP12 [32]. ORFs 2–7, at the 3' end of the genome, mainly encode glycoprotein (GP) 2, the envelope protein, GP3, GP4, GP5, ORF5a, the membrane matrix protein, and the nucleocapsid protein (N) [13]. Studies in China and abroad have shown that the variation seen in different strains of PRRSV is distributed across the genome, with a difference of up to 40% among them, but two genes, NSP2 and GP5, are the most variable [16]. On the basis of such differences in the viral genome sequences and the pathogenicity and antigenic characteristics of the virus, PRRSV falls into two major genotypes: the European type (genotype 1) and the North American type (genotype 2) [1, 10, 19]. The nucleotide sequence identity between these two genotypes is only approximately 60% [18, 20].
Since its first isolation in North America in 1987, PRRSV has been isolated in the Netherlands [29] and the USA [2], and it has impacted the global pig industry for nearly 40 years [21, 33]. In 2006, outbreaks of highly pathogenic (HP) PRRSV began to spread in China [27]. Clinically, PRRS causes high mortality in pig herds of all ages and also causes severe reproductive disorders in pregnant sows. The molecular characteristics of the HP-PRRSV genome include a unique 30 (1+29) discontinuous amino acid (aa) deletion in NSP2. The clinical features of this infection include pyrexia (40 °C–42 °C), a high incidence rate (50%–100%), and a high pathogenicity rate (20%–100%) [6]. Isolates representing the HP-PRRSV type include HUN4 and JXA1 [35]. That these HP strains are now predominant in China has seriously impacted China’s pig farming industry.
Many aspects of PRRSV, such as its pathogenic mechanisms, are not well understood. Therefore, establishing a reverse genetics system for this pathogen would help to clarify many outstanding issues at the molecular level. Many full-length infectious cDNA clones of RNA viruses have been successfully constructed [J et al., 1998, [22, 28, 31]. Most of the reverse genetics platforms for PRRSV are based on the classical European and North American PRRSV strains, and there have been few reports on infectious clones of HP-PRRSV strains [14, 36].
In this study, we successfully isolated a strain of HP-PRRSV and named it GDzj. We investigated the genetic relationships of GDzj by performing molecular evolutionary analysis and nucleotide sequence comparisons, and we examined its pathogenicity using challenge infections in piglets. We also constructed a full-length infectious cDNA clone of the GDzj strain using the bacterial artificial chromosome (BAC) system, providing an efficient platform for further research on the biological characteristics of this virus.
Materials and methods
Cells, clinical samples, strains and vectors
PRRSV cultures were passaged in Marc-145 cells. Lung samples were collected from sows on a commercial pig farm in Guangdong Province, China, that displayed clinical signs of PRRS. These sows had undergone stillbirths and showed signs of pyrexia, dyspnea, and spontaneous abortion. Trans5α competent cells and BAC vector plasmids (pSMART BAC v2.0; BamHI linearized) were preserved in the Institute of Swine Disease Control and Prevention, College of Animal Science, South China Agricultural University. BAC competent cells were purchased from Bomad Biotechnology (Gu'an, China) Co., Ltd. The pEASY-Blunt vector was purchased from Beijing TransGen Biotechnology (Beijing, China) Co., Ltd.
Main reagents
DNA and RNA extraction kits were purchased from Magen (Shanghai, China). Reverse transcription reagents, agarose gel DNA recovery kits, and plasmid extraction kits were all purchased from Promega (USA). High-fidelity DNA polymerase was purchased from Baori Biotechnology Co., Ltd. (Beijing, China) DS2000 and DS15000 DNA markers were purchased from Guangzhou Dongsheng Biotechnology Co., Ltd. Restriction endonucleases, T4 DNA ligase, Lipofectamine LTX, and associated reagents were purchased from Thermo Fisher Scientific (USA). Homologous recombinase and 2× Taq Plus Master Mix were purchased from Nanjing Novezin Biotechnology Co., Ltd. Fetal bovine serum, trypsin, DMEM high-glucose medium, and opti-MEM were purchased from GIBCO (USA). The PRRSV N protein monoclonal antibody was purchased from Beijing Yiqiao Shenzhou Technology Co., Ltd., and the Coralite 488-conjugated Affinipure Goat Anti-Rabbit IgG (H+L) secondary antibody was purchased from Guangzhou Guanjing Trading Co., Ltd.
Virus isolation and viral RNA extraction
The lung tissues with lesions used in this study were collected from a pig farm with suspected cases of PRRS in Guangdong Province. Individual lung tissues were cut into pieces, mixed with phosphate-buffered saline (PBS), and ground. After centrifugation, the supernatant was filtered through a 0.22-μm filter. Marc-145 cells were used to isolate PRRSV. When approximately 80% of the virus-infected cells showed a cytopathic effect (CPE), the virus was obtained from them by freeze-thawing and then passaged blindly on Marc-145 cells for three generations. Viral RNA was extracted in accordance with the instructions from the DNA/RNA extraction kit (Magen). The extracted RNA was amplified by reverse transcription polymerase chain reaction (RT-PCR) using NSP2 primers to confirm the presence of PRRSV, and by PCR testing to ensure that porcine epidemic diarrhea virus (PEDV) and porcine circovirus (PCV) 2 and 3 were not present (Table 1 and Fig. 1).

Detection of PRRSV, PEDV, PCV2, and PCV3. M, Marker DS2000 bp DNA; lanes 1–3, PRRSV NSP2 PCR products; lanes 4–6, PEDV ORF3 PCR products; lanes 7–9, PCV2 PCR products; lanes 10–12, PCV3 PCR products; lanes 2, 5, 8, and 11, positive control; lanes 3, 6, 9, and 12, negative control
The extracted viral RNA was reverse transcribed using a reverse transcription kit (Promega) with random primers, and each reaction included 6 μl of RNA template, 4 μl of reaction buffer, 2 μl of enzyme mix, and RNase-free water added to a total volume of 20 μl. The resulting cDNA was PCR-amplified according to the operating instructions for the high-fidelity DNA polymerase, and the purified PCR product was ligated to the pEAST-Blunt vector and used to transform Trans5α competent cells. Positive bacteria were selected for gene sequencing, and the sequencing products were automatically spliced together in silico using DNASTAR (USA) software to obtain the whole genome sequence of the virus. The complete genome sequence was named GDzj and was submitted to the GenBank database under the accession number MF772778.1.
Identification and sequence analysis of the isolated viruses
The virus was inoculated onto a monolayer of Marc-145 cells at a multiplicity of infection (MOI) of 0.1, and the cells were incubated at 37 °C with 5% CO2. These cells were fixed with 4% paraformaldehyde for 30 min at 48 hours postinfection (hpi), permeabilized using 0.5% Triton X-100, washed three times with PBS for 10 min at room temperature, and then incubated with anti-PRRSV-N-protein antibody diluted 1:500 at 4 °C. After again washing three times with PBS, the cells were incubated with Alexa Fluor 488–conjugated goat anti-mouse IgG at 37 °C for 1 h. Finally, after three washes, the cells were observed by fluorescence microscopy.
We used Lasergene 7.1 (USA) and MEGA 6.0 (USA) software to compare the sequences of the sequenced strains to those of representative strains from China and abroad. We constructed phylogenetic trees based on the aa sequences of the NSP2 and GP5 genes of the GDzj strain and reference strains. Gene homology analysis was carried out using SimPlot (USA) software using representative PRRSV sequences from the GenBank database (Table 2).
Animal experiments
To further study the pathogenic characteristics of the GDzj strain, an animal challenge experiment was performed using 21-day-old SPF weaned piglets. Laboratory tests for PRRSV, PCV2, PEDV, and Mycoplasma pneumoniae were all negative. The piglets, which were divided into two groups, were kept in separate spaces with separate water, food, and heating. One group (n = 3) was used as a control group, and the other group (n = 5) was used as the challenge infection group. Piglets in the control group were inoculated with 2 ml of porcine alveolar macrophage culture supernatant. Piglets in the infection challenge group were inoculated intranasally with 2 ml of the virus at a concentration of 105 TCID50/ml. The clinical effects of PRRSV infection were monitored, including changes in food intake, mental state, breathing, eyelid edema and secretions, exercise status, and skin and mucous membrane color. A separate thermometer was used to measure the rectal temperature of each pig once a day, and the weight of each pig was determined once every 3 days. Serum, which was collected from the piglets on days 0, 3, 5, 7, 10, 14, 17, and 21 post-challenge, was used to measure antibody and antigen levels and was also used for RT-qPCR to determine the viral load in each piglet.
During the experiments, piglet survival was recorded and a survival curve was drawn. Dead piglets were necropsied, and the remaining live piglets were euthanized at 21 dpi. To investigate tissue damage in the piglets and to determine the location of PRRSV in the lung tissues after challenge infection, the lungs from the piglets in each challenge group were collected, the regions showing obvious lesions were fixed, the tissues were sectioned, and immunohistochemical analysis was performed.
Construction of a full-length cDNA infectious clone of PRRSV
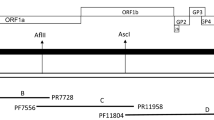
The scheme used for construction of a full-length cDNA clone of the GDzj strain is shown in Fig. 2. Briefly, the construction involved transforming competent cells with a low-copy-number BAC plasmid, designing and synthesizing a gene containing a CMV promoter, a multi-enzyme cleavage site, a hepatitis D virus ribozyme, and a BGH termination signal sequence, as shown in Fig. 2. The construct was ligated to the BAC plasmid via restriction enzyme digestion and ligation, and positive plasmids were identified by screening. A recombinant plasmid verified by sequencing was named pBAC.

pBAC-GDzj plasmid construction strategy. The fragment that includes the unique restriction enzyme site is shown in the illustration. The four overlapping fragments from the full-length GDzj genome produced by PCR amplification are shown in the middle section.
The cDNA obtained by reverse transcription was amplified by RT-PCR using the primers shown in Table 3, and four fragments were obtained. The fusion PCR method was used to mutate position 3407 in the GDzj genome, resulting in a cysteine-to-alanine mutation and introducing a single restriction site for BstBI as a genetic marker for rescuing the virus. The amplified PCR products were recovered and purified separately, and the pBAC plasmid was digested using the restriction sites shown in Fig. 2. Four fragments were obtained, and fragments Bzj1 and Bzj4 contained portions of the pBAC plasmid. After double digestion of the pBAC vector with the appropriate restriction enzymes, the fragment Bzj1 was inserted into the pBAC vector using a homologous recombination kit according to the instructions, and the positive pBAC-Bzj1 plasmid was confirmed by sequencing. The other three fragments were inserted sequentially into the plasmid in the same way, resulting in a full-length cDNA plasmid clone, which was confirmed by sequencing and named pBAC-GDzj.
Rescue and identification of the virus
Marc-145 cells were transfected with the pBAC-GDzj plasmid when the cell density reached 70%–80%, using Lipofectamine LTX. At 96 h post-transfection, 90% of the cells contained lesions. After three repeated freeze-thaw cycles, the cell culture medium was collected and inoculated onto a monolayer of Marc-145 cells. After adsorption at 37°C for 1 h, and the supernatant was discarded, the cells were washed three times with PBS buffer, DMEM cell maintenance medium containing 2% fetal bovine serum (FBS) was added, and the plate was placed in an incubator at 37°C with 5% CO2. The cells were examined daily for CPE. Approximately 90% of the lesion-containing regions were freeze-thawed to harvest the virus. The rescued virus was named RvGDzj. The experiment was set up with the parental GDzj virus and untransfected Marc-145 cells as controls.
Indirect immunofluorescence assay (IFA), RT-PCR, single enzyme digestion identification, and sequence determination were used to identify the third-generation (P3) rescued virus. The third-generation RvGDzj-P3 (from the rescued virus) and the third-generation GDzj-P3 (from the parental virus) were inoculated separately onto a Marc-145 cell monolayer, and the plate was placed in an incubator at 37°C with 5% CO2. After 48 h, IFA was conducted in accordance with standard procedures, and a PRRSV N protein antibody was used for IFA detection. For RT-PCR identification, 200 μL of the rescued virus suspension was used for viral RNA extraction using a DNA/RNA extraction kit, and RT-PCR amplification was performed with PRRSV GP5- and NSP2-specific primers (Table 4). The amplified PCR products were subjected to agarose gel electrophoresis and then sequenced. In these experiments, the parental GDzj virus and untransfected Marc-145 cells were used as positive and negative controls, respectively.
Virus growth kinetics
Growth curves for the third-generation RvGDzj virus and the third-generation parental GDzj virus were determined using Marc-145 cells. Marc-145 cells were pre-cultured in a 12-well cell culture plate to obtain a monolayer, and the third-generation RvGDzj virus and the third-generation GDzj parental virus were inoculated separately at an MOI of 0.1 onto the cell monolayer and incubated in an incubator at 37°C with 5% CO2 for 1 h. Three replicates were performed for each virus. After incubation, the supernatant was discarded, the cells were washed three times with PBS buffer, DMEM cell maintenance medium containing 2% FBS was added, and the cells were cultured in an incubator at 37°C with 5% CO2. The supernatant was collected at 12, 24, 36, 48, 60, and 72 hpi.
Marc-145 cells were seeded onto a 96-well cell culture plate. When the cell confluence exceeded 90%, the rescued virus obtained was diluted in a tenfold series. For each dilution, four wells of a plate seeded with Marc-145 cells were inoculated separately with either virus (100 μL per well), and the blank control was not inoculated. The cell culture plate was incubated at 37 °C with 5% CO2. The cells were observed continuously for 2–7 days, the number of cytopathic wells at each dilution was determined, and the 50% tissue culture infectious dose (TCID50) was calculated for the viral supernatant. Finally, the infection time was used as the abscissa, and the TCID50 measured at this time point was used as the ordinate for drawing a growth curve for the virus, which enabled the growth characteristics of the rescued and parental viruses to be compared in Marc-145 cells.
Statistical analysis
Numerical data are expressed as the mean ± SD and were analyzed using GraphPad Prism software (version 8.0.1 for Windows; GraphPad Software Inc.). Differences between groups were assessed using ANOVA. Differences were considered statistically significant when the P-value was < 0.05 and extremely significant when the P-value was < 0.01 or P < 0.001.
Results
Isolation and identification of viruses
Obvious cytopathic changes were observed microscopically after the diseased material was inoculated onto Marc-145 cells for 72 hours, whereas the uninfected Marc-145 control cells showed no such changes (Fig. 3A and B). IFA showed that the virus-inoculated cells were observable by fluorescence microscopy. A lack of green fluorescence in the blank Marc-145 control cells showed that they did not specifically react with the anti-PRRSV N protein antibody, thereby confirming that virus isolation was successful (Fig. 3C and D).

Virus isolation and identification. (A and B). Cytopathic effect in the Marc-145 cells infected with GDzj at 72 hours post-infection (hpi). (C and D). Indirect immunofluorescence of GDzj-infected and mock‐infected Marc-145 cells at 72 hpi using a monoclonal antibody against the PRRSV N protein.
Sequence analysis
We used DNASTAR 7.1 software to analyze the whole genome of the GDzj strain and representative strains from China and abroad and constructed an evolutionary tree from the data. The results showed that the GDzj strain belongs to lineage 8.7 (represented by the JXA1-like strain) and shares the highest nucleotide sequence similarity with HUN4; therefore, GDzj is an HP-PRRSV strain (Fig. 4A). The sequence of the whole genome of the GDzj strain is 58.5% identical to that of the representative European LV strain, 88.4% identical to that of the representative North American VR-2332 strain, and 98.2% and 97.9% identical to those of the Chinese domestic HUN4 and JXA1 strains, respectively (Fig. 4B). This shows that the GDzj strain belongs to the North American type and is closely related to the HUN4 strain and the JXA1 strain. The levels of the whole gene sequences from the GDzj strain and the reference JXA1 and HUN4 strains (Fig. 4C) indicate that GDzj is an HP strain.



Phylogenetic analysis (A) nucleotide sequence comparisons (B), and homologous recombination analysis (C) of whole viral genome sequences. The similarity plot was generated using GDzj as the query sequence against those of JXA1 (blue) and HUN4 (red). (C and E) Amino acid sequence alignment and evolutionary tree analyses of the GP5 (F) and NSP2 (G) genes of strain GDzj. The phylogenetic tree was constructed by the neighbor-joining method using Lasergene 7.1 and MEGA 6.06 software. The genome sequence comparisons were conducted using SimPlot 3.51 software.
The NSP2 and GP5 sequences from the GDzj strain were further analyzed and compared with those of the reference strain. We identified two non-contiguous aa deletions in the NSP2 gene of GDzj, a finding that is consistent with the known deletion sites in the HP strain (Fig. 4D). The GP5 gene contains an N34G mutation at position 34, an I94A mutation at position 94, and a Q196R mutation at position 196 (Fig. 4E). Based on these genetic differences, our aa-based phylogenetic tree showed that NSP2 and GP5 belong to the branch represented by the HP strains HuN4, JXA1, and JXwn06 (Fig. 4F and G).
Clinical manifestations in piglets and their lung and hilar lymph node characteristics in PRRSV challenge experiments
In our challenge infections, from the second day of the challenge, piglets in the experimental group appeared to have messy fur, a depressed mood, and decreased food intake, accompanied by temperature rise and fever. In the later part of the challenge infections, some piglets died and the body temperatures of some of them dropped slightly.
The mean body temperature of the GDzj challenge group was significantly higher than that of the control group after challenge, with the highest temperature reaching 41.5°C, a fever state (Fig. 5A). The score relating to the clinical signs in the GDzj challenge group was significantly higher than that of the control group (Fig. 5B), the weight gain was significantly lower than that of the control group (Fig. 5C), and the mortality rate reached 40% (Fig. 5D).

Clinical scores and post-mortem tissue changes in the experimental animals after virus challenge. (A) Rectal temperatures recorded for 21 days after virus challenge. Data are the mean ± standard deviation (error bars). (B) Clinical scores after virus challenge. Data were recorded as described previously [15], and the mean values were calculated on days 1–3, 4–5, 6–7, 8–10, 11–14, 15–17, and 18–21 postinfection (p.i.). Asterisks indicate significant differences between the GDzj-inoculated group and the control group (*, p < 0.05; **, p < 0.01; ***, p < 0.001). (C) Average daily weight gain in the laboratory animals after viral challenge. Data were recorded and the mean values were calculated on days 1, 3, 5, 7, 10, 14, 17, and 21 p.i. (D) Survival of laboratory animals after virus challenge. (E and F) Serum antigen (E) and antibody (F) status of the experimental animals after infection challenge. (G) Lungs and hilar lymph nodes of infected and uninfected animals. (H) Pathological sections in the lungs from the experimental animals after viral challenge. Lung sections were stained with hematoxylin and eosin (H & E) and also examined by immunohistochemistry (IHC) using monoclonal antibodies specific for the N protein of PRRSV.
After the challenge infections, serum antibodies and antigen levels in the GDzj group and the control group were measured. In the GDzj group, antibodies became detectable at 7 days after challenge, and the antibody level gradually increased and antigen became detectable at 1 day after challenge (Fig. 5E). Serum antigen levels in the GDzj group were highest at 7 days post-challenge but then gradually decreased and stabilized. Piglets in the control group were negative for both antigen and antibodies (Fig. 5F).
The lungs from the piglets in the challenge group showed obvious edema, with "meat-like changes" occurring in many places and being almost diffuse throughout the lungs, and the interlobular interval was significantly widened, showing the typical signs of interstitial pneumonia (Fig. 5G). Analysis of the prepared pathological tissue sections from the autopsied material showed that the lung tissue from the piglets in the infection challenge group showed obvious signs of pneumonia, as mainly manifested by thickening of the alveolar septum, a reduction in the alveolar cavity volume, and proliferation of inflammatory cells. The prepared immunohistochemical sections showed obvious tan-colored positive signals in the lung tissues from the piglets in the challenge group, and tracer virus particles were mainly located in alveolar macrophages and epithelial cells, but these signals were not seen in the control group (Fig. 5H).
Rescue and virus identification
Marc-145 cells were transfected with the full-length cDNA pBAC-GDzj plasmid, and after 96 hours, 90% of the cells displayed cytopathic changes and the virus was harvested by freeze-thawing. This virus suspension was inoculated onto Marc-145 cells, and CPE was observed. The results showed that, microscopically, the CPE was similar to that seen with the parental GDzj virus, and the rescued virus was named RvGDzj. As shown in Fig. 6A, compared with the control cells (i.e., uninfected Marc-145 cells), the cells inoculated with the rescued virus or the parental virus showed evidence of shrinkage and accumulated protrusions, whereas the control cells always appeared normal.

Rescue and identification of viruses. (A) Cytopathic effects in Marc-145 cells infected with the rescued viruses at 36 hpi. (B) PCR identification of the rescued virus. M, Marker DS2000 bp DNA; lane 1, GP5 fragment from the rescued virus (RvGDzj); lane 2, GP5 fragment from the parent virus (GDzj); lane 3, negative control; lane 4, NSP2 fragment from the rescued virus (RvGDzj); lane 5, NSP2 fragment from the parent virus (GDzj); lane 6, negative control. Restriction enzyme digestion, IFA, and western blotting were used to identify the rescued virus (RvGDzj). (C) Restriction enzyme digestion and identification of PCR products from the rescued viruses. M, Marker DS2000bp DNA, lane 1, restriction fragment from the rescued virus (RvGDzj); lane 2, restriction fragment from the parent virus (GDzj). (D) Western blot identification of the rescued virus (RvGDzj). (E) IFA identification of the rescued virus
The rescued RvGDzj was serially passaged in Marc-145 cells. We took the culture medium from the third-generation rescued virus (RvGDzj-P3), extracted RNA from it, and used PRRSV GP5 and NSP2 primers for RT-PCR tests. Upon agarose gel electrophoresis, a strong PCR amplicon was detected. Sequencing of this amplicon showed that the GP5 and NSP2 sequences from the rescued virus were identical to those of the parent strain (Fig. 6B).
To distinguish the parental virus from the rescued virus, genome position 3407 was mutated, a BstBI restriction endonuclease site was added for cloning purposes, and the rescued virus fragment was amplified by RT-PCR. BstBI was used to distinguish the two viruses by restriction digestion, and two fragments were only obtained from the rescued virus (Fig. 5C).
A monoclonal antibody targeting the PRRSV N protein was used in IFA to distinguish cells infected with the third generation of the rescued virus (RvGDzj-P3) from mock-infected cells (Fig. 6D). Marc-145 cells inoculated with the parental virus or with the rescued virus displayed the specific green fluorescence characteristic of anti-PRRSV N-protein positivity under a fluorescence microscope, whereas the blank control cells did not, which shows that the virus rescue was successful and that the rescued virus was recognized by this antibody (Fig. 6E).
Comparison of the in vitro growth kinetics of the parental strain and the rescued virus
The growth curves of the third-generation rescued virus (RvGDzj-P3) and the third-generation parental virus (GDzj-P3) were compared and found to be similar. However, the viral titer of the parental virus reached a peak at 36 hpi and decreased thereafter, while that of the rescued virus peaked at 72 hpi with a titer higher than at 12, 24, 60, and 72 hpi, but the difference was not significant (Fig. 7).

Growth curves for the rescued and parental viruses. Marc-145 cells were infected with GDzj or RvGDzj at a MOI of 0.1. Supernatants from the infected cells were collected at the time points indicated (12, 24, 36, 48, 60, and 72 hpi). Viral titers were determined by a TCID50 assay in Marc-145 cells and used to produce growth curves.
Discussion
PRRSV is a major disease afflicting the global swine industry. Despite PRRSV becoming more prevalent over the last 35 years, it has not yet been substantively controlled globally, so its economic impact and harm to the swine industry remain unchanged. PRRSV is an RNA virus with an extremely high mutation rate and propensity for fast evolution. The increasing diversity of strains and the increasing frequency of the appearance of new strains and recombinant strains have made its prevention and control difficult [5, 9, 25]. The in-depth study of this virus largely depends on the ability to construct infectious virus clones. In 2006, an outbreak of PRRSV caused by HP-PRRSV occurred in China. Some laboratories have isolated several HP-PRRSV strains and established reverse genetic systems [7, 8, 17, 24].
Reverse genetics is a virus rescue technology that copies the virus from a cDNA clone of theviral genome. The strategy to rescue infectious viruses from plasmids encoding a cloned cDNA corresponding to the viral genome is one of the most important technological advances in experimental virology systems. As a technology platform, it can be used to study the genome structure, gene functions, and pathogenic mechanisms of RNA viruses [3]. As one of the basic technology platforms employed by many molecular biology laboratories, reverse genetics technology has been widely used in various fields of life science research. Its application scope includes not only the study of gene replication and regulation of gene expression in animal viruses and the interactions occurring between viruses and their hosts but also research on antiviral strategies and gene therapy, the development of new genetically engineered vaccines, and the construction of new target viral vectors [34]. This technology can also provide a basic technical resource for the prevention and control of RNA viruses causing major diseases in various animals, such as African swine fever, peste des petits ruminants, and Schmallenberg disease [11, 12, 23].
In the process of constructing infectious clones of PRRSV, early research mainly adopted the establishment of a virus rescue system by placing clones containing the full-length cDNA of the PRRSV genome under a prokaryotic transcription promoter such as a phage T7 or SP6 promoter. The necessity to generate full-length RNA PRRSV transcripts by in vitro transcription, however, means that this process has many operational limitations. To circumvent this, we combined a BAC-based system with homologous recombination technology to construct a full-length cDNA infectious clone of PRRSV. This system offers several advantages over similar systems. First, because the BAC plasmid can accommodate up to 350 kb of foreign DNA, it can be used to clone large foreign genes. It is also suitable for the construction of infectious clones of viruses with large genomes. Second, CMV is widely recognized as the most powerful promoter for initiating eukaryotic gene expression. A BAC construct made by placing the clone of the full-length viral genome under the control of the CMV promoter can be used directly to transfect eukaryotic cells without in vitro transcription, thereby saving time. Third, the use of homologous recombination technology eliminates the need for restriction enzyme sites, greatly improving the efficiency of construction of full-length cDNA clones and reducing costs in the process.
The ability to construct full-length infectious cDNA clones mainly hinges on whether a faithful whole-genome fragment can be obtained for the virus of interest. In the case of PRRSV, because its genome is relatively large, obtaining a full-length cDNA sequence is difficult. Therefore, we avoided introducing gene mutations and deletions during the construction of the full-length infectious cDNA clones by dividing the whole genome from the HP-PRRSV GDzj strain into four fragments during the construction process. High-fidelity enzymes were used for amplification to reduce the number of mutations during the RT-PCR process. To ensure the fidelity of the constructed infectious molecular clone, we also sequenced the full length of the plasmid. Thus, we successfully constructed a full-length infectious clone from the GDzj strain, and a virus (RvGDzj) was rescued. It is worth noting also that in the present study the rescued recombinant PRRSV and the parental virus displayed similar proliferation ability in vitro. This infectious clone thus provides a powerful tool for further investigating the pathogenesis of PRRSV.
In summary, we isolated a PRRSV strain GDzj from lung tissue from a pig with suspected PRRS infection. In experimental challenge infections of weaned piglets, this virus caused severe fever, difficulty in breathing, loss of appetite, and other clinical manifestations in the piglets and some infections were fatal. Necropsy revealed the presence of lesions in the lung tissue and hilar lymph nodes. Our next experiments, which involved constructing a full-length cDNA infectious clone of GDzj using the BAC system, showed that the rescued virus displayed in vitro proliferation characteristics similar to those of the parental PRRSV. The outcome of our research not only provides a useful reverse genetics platform for exploring the pathogenesis of HP-PRRSV but also lays a foundation for exploring and studying other RNA viruses with even larger genomes.
Availability of data and materials
Genetic data presented in this paper are publicly available via GenBank.
Abbreviations
- HP-PRRSV:
-
Highly pathogenic porcine reproductive and respiratory syndrome virus
- PRRS:
-
Porcine reproductive and respiratory syndrome
- ORFs:
-
Amino acid, aa; open reading frame
- NSP:
-
Non-structural protein
- BAC:
-
Bacterial artificial chromosome
- RT-PCR:
-
Reverse transcription polymerase chain reaction
- MOI:
-
Multiplicity of infection
- MCS:
-
Multiple cloning site
- FBS:
-
Fetal bovine serum
- IFA:
-
Immunofluorescence assay
References
Adams MJ, Lefkowitz EJ, King AM et al (2016) Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses. Arch Virol 161:2921–2949
Benfield DA, Nelson E, Collins JE et al (1992) Characterization of swine infertility and respiratory syndrome (SIRS) virus (isolate ATCC VR-2332). J Vet Diagn Invest 4:127–133
Boyer JC, Haenni AL (1994) Infectious transcripts and cDNA clones of RNA viruses. Virology 198:415–426
Cavanagh D (1997) Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch Virol 142:629–633
Chaikhumwang P, Tantituvanont A, Tripipat T et al (2015) Dynamics and evolution of highly pathogenic porcine reproductive and respiratory syndrome virus following its introduction into a herd concurrently infected with both types 1 and 2. Infect Genet Evol 30:164–174
Chen N, Xiao Y, Ye M et al (2020) High genetic diversity of Chinese porcine reproductive and respiratory syndrome viruses from 2016 to 2019. Res Vet Sci 131:38–42
Fang Y, Faaberg K S, Rowland R R R, et al (2006a) Construction of a full-length cDNA infectious clone of a European-like type 1 PRRSV isolated in the US. Nidoviruses: Toward Control of Sars and Other Nidovirus Diseases, 581: 605–608
Fang Y, Rowland RRR, Roof M et al (2006) A full-length cDNA infectious clone of North American type 1 porcine reproductive and respiratory syndrome virus: expression of green fluorescent protein in the Nsp2 region. J Virol 80:11447–11455
Feng YJ, Zhao TZ, Nguyen T et al (2008) Porcine respiratory and reproductive syndrome virus variants, Vietnam and China, 2007. Emerg Infect Dis 14:1774–1776
Forsberg R, Storgaard T, Nielsen HS et al (2002) The genetic diversity of European type PRRSV is similar to that of the North American type but is geographically skewed within Europe. Virology 299:38–47
Hu HX, Roth JP, Yu QZ (2018) Generation of a recombinant Newcastle disease virus expressing two foreign genes for use as a multivalent vaccine and gene therapy vector. Vaccine 36:4846–4850
Huang ZH, Elankumaran S, Yunus AS et al (2004) A recombinant newcastle disease virus (NDV) expressing VP2 protein of infectious bursal disease virus (IBDV) protects against NDV and IBDV. J Virol 78:10054–10063
Johnson CR, Griggs TF, Gnanandarajah J et al (2011) Novel structural protein in porcine reproductive and respiratory syndrome virus encoded by an alternative ORF5 present in all arteriviruses. J Gen Virol 92:1107–1116
Kwon B, Ansari IH, Pattnaik AK et al (2008) Identification of virulence determinants of porcine reproductive and respiratory syndrome virus through construction of chimeric clones. Virology 380:371–378
Li Y, Zhou L, Zhang J et al (2014) Nsp9 and Nsp10 contribute to the fatal virulence of highly pathogenic porcine reproductive and respiratory syndrome virus emerging in China. PLoS Pathog 10:e1004216
Liu JK, Wei CH, Yang XY et al (2013) Genetic diversity and evolutionary characterization of Chinese porcine reproductive and respiratory syndrome viruses based on NSP2 and ORF5. Arch Virol 158:1811–1816
Lv J, Zhan JW, Sun Z et al (2008) An infectious cDNA clone of a highly pathogenic porcine reproductive and respiratory syndrome virus variant associated with porcine high fever syndrome. J Gen Virol 89:2075–2079
Murtaugh MP, Elam MR, Kakach LT (1995) Comparison of the structural protein coding sequences of the VR-2332 and Lelystad virus strains of the PRRS virus. Arch Virol 140:1451–1460
Murtaugh MP, Stadejek T, Abrahante JE et al (2010) The ever-expanding diversity of porcine reproductive and respiratory syndrome virus. Virus Res 154:18–30
Nelsen CJ, Murtaugh MP, Faaberg KS (1999) Porcine reproductive and respiratory syndrome virus comparison: divergent evolution on two continents. J Virol 73:270–280
Neumann EJ, Kliebenstein JB, Johnson CD et al (2005) Assessment of the economic impact of porcine reproductive and respiratory syndrome on swine production in the United States. J Am Vet Med Assoc 227:385–392
Nielsen HS, Liu G, Nielsen J et al (2003) Generation of an infectious clone of VR-2332, a highly virulent North American type isolate of porcine reproductive and respiratory syndrome virus. J Virol 77:3702–3711
Pollin R, Granzow H, Kollner B et al (2013) Membrane and inclusion body targeting of lyssavirus matrix proteins. Cell Microbiol 15:200–212
Ran ZG, Chen XY, Yang HC et al (2007) Recovery of an infectious virus from the full-length cDNA of PRRSV BJ-4. Wei Sheng Wu Xue Bao 47:423–429
Saenglub W, Jantafong T, Mungkundar C et al (2020) Genetic signatures of the immune-escaping type 2 porcine reproductive and respiratory syndrome virus in farms with a robust vaccination program. Microb Pathog 144:104166
Snijder EJ, Meulenberg JJ (1998) The molecular biology of arteriviruses. J Gen Virol 79(Pt 5):961–979
Tian KG, Yu XL, Zhao TZ et al (2007) Emergence of fatal PRRSV variants: unparalleled outbreaks of atypical PRRS in China and molecular dissection of the unique hallmark. PLoS ONE 2:e526
Wang J, Duan J, ZhU L et al (2015) Sequencing and generation of an infectious clone of the pathogenic goose parvovirus strain LH. Arch Virol 160:711–718
Wensvoort G, Terpstra C, Pol JM et al (1991) Mystery swine disease in the netherlands: the isolation of Lelystad virus. Vet Q 13:121–130
Wootton S, Yoo D, Rogan D (2000) Full-length sequence of a Canadian porcine reproductive and respiratory syndrome virus (PRRSV) isolate. Arch Virol 145:2297–2323
Xi J, Zhang Y, Wang J et al (2017) Generation of an infectious clone of AMDV and identification of capsid residues essential for infectivity in cell culture. Virus Res 242:58–65
Xu H, Song S, Zhao J et al (2020) A potential endemic strain in China: NADC34-like porcine reproductive and respiratory syndrome virus. Transbound Emerg Dis
Yang S, Oh T, Kang I et al (2019) Efficacy of concurrent vaccination with modified-live PRRSV-1 and PRRSV-2 vaccines against heterologous dual PRRSV-1 and PRRSV-2 challenge in late term pregnancy gilts. Vet Microbiol 239:108497
Zhang S, Zhou Y, Jiang Y et al (2011) Generation of an infectious clone of HuN4-F112, an attenuated live vaccine strain of porcine reproductive and respiratory syndrome virus. Virol J 8:410
Zhou L, Yang H (2010) Porcine reproductive and respiratory syndrome in China. Virus Res 154:31–37
Zhou L, Zhang J, Zeng J et al (2009) The 30-amino-acid deletion in the Nsp2 of highly pathogenic porcine reproductive and respiratory syndrome virus emerging in China is not related to its virulence. J Virol 83:5156–5167
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2018YFD0501100), Major animal disease prevention and control technology research and promotion projects (201817SY0002), Guangdong Province Key Field R&D Program Project (2019B020211003), The 2020 Youth Backbone Training Plan of Henan University of Higher Education (GaoJiao[2020]354), Key and Cultivation Discipline of Xinyang Agriculture and Forestry University (ZDXK201702), We thank Sandra Cheesman, PhD, from Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Funding
This work was supported by the National Key Research and Development Program of China (2018YFD0501100), Major animal disease prevention and control technology research and promotion projects (201817SY0002), Guangdong Province Key Field R&D Program Project (2019B020211003), The 2020 Youth Backbone Training Plan of Henan University of Higher Education (GaoJiao[2020]354), Key and Cultivation Discipline of Xinyang Agriculture and Forestry University (ZDXK201702)
Author information
Authors and Affiliations
Contributions
SW, JD, and LY contributed equally to the work; SW, LY, and PZ performed the assays and performed the data analyses; TL, LZ, PL, and LW performed the sequence alignment; and JD, ZX and CS wrote the manuscript. All authors reviewed and approved the final form of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval and consent to participate
All of the samples were collected according to the animal ethics regulations of the National Engineering Center for Swine Breeding Industry (NECSBI 2015-16).
Additional information
Handling Editor: Jens H. Kuhn.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wang, S., Liu, Y., Yu, L. et al. A strain of highly pathogenic porcine reproductive and respiratory syndrome virus: genomic characterization, pathogenicity, and construction of an infectious full-length cDNA clone. Arch Virol 166, 3127–3141 (2021). https://doi.org/10.1007/s00705-021-05212-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-021-05212-w