
Abstract
To investigate canine kobuvirus (CaKoV) infection in southwest China, 107 fecal samples were collected from dogs with obvious diarrhea in Sichuan and Chongqing regions, China. CaKoV infection was detected in 54 diarrheic samples (50.46%) by RT-PCR targeting a partial fragment (504 bp) of the 3D gene. Comparison of these partial 3D gene sequences from 14 of these CaKoV-positive samples show 95.4%–99.0% nucleotide (nt) identity within this group, and nt identities ranging from 93.1% to 98.2% with previously reported CaKoV 3D gene sequences. Additionally, we amplified five VP1 gene sequences and analyzed the inferred phylogeny. Amino acid (aa) identities of the five VP1 gene sequences were 81.5%–89.4% with those previously reported. Furthermore, one complete CaKoV genome was successfully obtained from a positive sample and designated SMCD-59/CHN/2015. This genome consisted of 8,184 nt, and shared 92.9%–96.6% nt identity (97.6%–98.1% aa identity) with other reported CaKoV genomes. This study provides proof that CaKoV circulates in diarrheic dogs in southwest China, and that these viruses exhibit unique genetic characteristics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Canine kobuvirus (CaKoV) is classified as a novel member of the Aichivirus A species, which taxonomically belongs to the Kobuvirus genus in the family Picornaviridae [1]. According to previous studies, the CaKoV genome is 8.1–8.2 kb long, with a single open reading frame (ORF) containing 7,332–7,341 nucleotides (nt), encoding 2,442–2,475 amino acids (aa) [2, 3]. All CaKoV isolates have the same genomic organization: a 5′-untranslated region (UTR); a single ORF encoding a polyprotein precursor that is processed to generate a leader (L) protein, three structural proteins (VP0, VP3, and VP1), and seven non-structural proteins (2A, 2B, 2C, 3A, 3B, 3C, and 3D); a 3′-UTR; and a poly-(A) tail [1]. CaKoV structural proteins form the virion capsid, which is associated with the adsorption and invasion of virus particles. CaKoV nonstructural proteins and intermediates are involved in RNA replication, polyprotein cleavage, and assembly of virions [1].
CaKoV was first detected in 2011 in the feces of dogs with acute gastroenteritis in the USA [2, 3]. CaKoV has subsequently been detected in diarrheic as well as healthy dogs in Britain [4], Italy [5], South Korea [6, 7], Tanzania and Kenya [8], Japan [9], and China [10, 11], presenting a global distribution. The emerging virus has been detected in wild carnivores, including jackal, fox, and Hyaenidae [8, 12], in addition to domestic dogs. Previous studies have shown no significant difference in CaKoV prevalence between healthy versus diarrheic dog samples [6]. Therefore, it is unclear whether CaKoV alone, or together with other canine enteric pathogens causes gastroenteritis in dogs. Recently, the first report of CaKoV being detected in extra-intestinal tissue was published; the virus was found within the cerebellum, lung, tonsil, and liver of a 5-month-old puppy [13], suggesting that CaKoV may lead to systemic infection. In this study we report the prevalence of CaKoV in diarrheic dog feces from southwest China, and we characterize the genome of SMCD-59/CHN/2015, a Chinese CaKoV strain.
Materials and methods
Sample collection
Fecal samples were collected from single-household dogs with diarrhea symptoms at six animal hospitals in Sichuan and Chongqing regions, China, during 2015 and 2016. The dogs were 2–6 months old, and were kept indoors, except for outdoor walks or runs of 1–2 hours daily. Labrador retriever, golden malinois, Tibetan mastiff, chihuahua, pomerania, schnauzer, and poodle breeds were all present. The samples were suspended in 5.0-mL sterile phosphate buffer saline (PBS) in sampling tubes, and then stored at −80°C for further testing.
RNA extraction and cDNA synthesis
Fecal samples were resuspended by vigorous vortexing in PBS at approximately 0.2 g/mL. The suspensions were centrifuged at 12,000 xg for 5 min to remove cellular debris. Viral RNA was extracted from 300 µL of the fecal suspensions using RNAios Plus® (TaKaRa BIO INC, JPN) according to the manufacturer’s instructions, and then reverse transcribed into cDNA using the PrimeScript™ RT Reagent Kit according to the manufacturer’s instructions (TaKaRa BIO INC, JPN). The resulting cDNA was used as a template for the PCR assay, and stored at −20 °C.
Detection of CaKoV
CaKoV RNA was detected by an RT-PCR assay targeting a 504 bp fragment of the 3D gene, with forward, CaKV/F (5′-CCCTGGAACACCCAAGGCCGCT-3′), and reverse primer, CaKV/R (5′-TCTGGTTGCCATAGATGTGGTG-3′), sequences, as described by Di Martino et al. [5]. PCR products were purified and cloned into the pMD19-T (TaKaRa BIO INC, JPN) simple vector, prior to sequencing. Sequences have been submitted to GenBank under accession No. MF062159–MF062171.
VP1 gene sequence amplification
The CaKoV VP1 gene was also analyzed. A primer pair was designed, according to the genome sequence (JQ11763) available in GenBank, to amplify the VP1 capsid-coding sequences from positive samples (VP1/F: 5′-GTGGACATCTTCAACCCTGAT-3′ and VP1/R: 5′-CCCACGGTTTGCCGAGTT-3′). PCR conditions were as follows: 94 °C for 2 min, followed by 40 cycles at 94 °C for 45 s, 54 °C for 45 s and 72 °C for 70 s, with a final extension step at 72 °C for 10 min. PCR products were 1,188-bp long, and contained the full VP1 gene sequence. After the amplification, products were purified and cloned into the pMD19-T simple vector, and then sequenced. VP1 sequences have been submitted to GenBank under accession No. MF062172–MF062175.
CaKoV genome amplification and phylogenetic analysis
Nine pairs of sequence-specific oligonucleotide primers were designed using the conserved regions of two published CaKoV sequences (JQ911763 and KF924623) to determine the CaKoV genome sequence. PCR reaction conditions are shown in Table 1. PCR products were purified and cloned into the pMD19-T simple vector, prior to sequencing. The sequences were assembled using SeqMan software (version 7.0; DNASTAR). The single ORF was identified using an online ORF finder (https://www.ncbi.nlm.nih.gov/orffinder/). The CaKoV genome sequence generated in this study has been deposited in GenBank under accession No. MF062158.
Sequence, recombination, and phylogenetic analyses
All sequence identity analyses were performed with the nt and aa sequences aligned with the ClustalW method using the MegAlign 7.2 program in the Lasergene software (DNASTAR, Madison, WI, USA). Neighbor-joining phylogenetic trees were built with the p-distance model, 1,000 bootstrap replicates and, otherwise, the default parameters in MEGA 6. The Recombination Detection Program version 4.0 (RDP) was used to identify possible parental sequences and recombinant strains.
Results
Detection of CaKoV
Of the 107 samples evaluated, 54 (50.46%) were positive for CaKoV. CaKoV positive rates were 50.8% and 50.0% in Sichuan and Chongqing regions, respectively, which is higher than in northeast China [10].
Partial 3D gene sequence and phylogenetic analyses
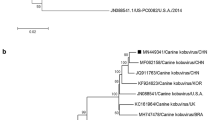
In this study 14 CaKoV partial 3D genes (504 nt) were sequenced, of the 54 positive samples identified. The 14 sequences shared 95.4%–99.4% nt identity with each other, and 94.1%–98.2% nt identity with other sequences previously reported. Phylogenetic analysis indicates that the 14 CaKoV sequences in this study are more closely related to other CaKoV sequences from Asia and Europe, than with sequences from Africa and the Americas (Fig. 1), which is consistent with previous reports [5, 10]. Moreover, the 14 Chinese CaKoV sequences cluster into two major groups, albeit with little bootstrap support. Interestingly, one sequence (SMCQ-S6) clusters alone, with the remaining 13 sequences clustering into several different, loosely supported for the most part, groups, along with the other sequences previously reported. Six of the 13 sequences appear more closely related to the CaKoV strains from South Korea and England, whereas seven of the 13 sequences seem to be more closely related to the CaKoV strains from China and Japan.

A neighbor-joining phylogenetic tree based on a sequence alignment of 504 nucleotides of the 3D gene from CaKoV strains; isolates from this study are marked with black boxes. Bootstrap values based on 1,000 replicates are shown at the nodes
VP1 genes sequence analysis
We obtained five CaKoV complete VP1 gene sequences in this study, ranging in length between 835–838 bp, and encoding 278–279 aa. Nt and deduced aa identities among these five CaKoV VP1 gene sequences range from 95.2% to 99.5% and 94.7% to 98.6%, respectively. Nt and aa identities are 83.0%–91.2% and 82.3%–89.7%, respectively, between these five CaKoV VP1 gene sequences and other CaKoV VP1 sequences previously reported. No reorganization of the gene was found using RDP software.
Phylogenetic analysis based on our and other reported CaKoV VP1 aa sequences divides the virus isolates into two major groups: one group of sequences from four sources, the United States, England, Korea, and Africa; and another group of sequences from China (Fig. 2). However, one Chinese sequence, JQ11763/CH-1, is not strongly associated with either cluster. Our five Chinese VP1 aa sequences cluster into a single group, with 100% bootstrap support, but the relationship with strain JQ11763/CH-1 remains unclear based on VP1 sequences. These five VP1 sequences share nine identical aa substitution sites, compared with the other six aa sequences previously reported, all concentrated at positions 201–243 (Table 2).

A neighbor-joining phylogenetic tree based on a sequence alignment of 279 amino acids inferred from the VP1 gene of CaKoV strains; isolates from this study are marked with black boxes. Bootstrap values based on 1,000 replicates are shown at the nodes
SMCD-59/CHN/2015 genome analysis
We amplified, using nine pairs of primers, sequenced, and assembled the genome of the SMCD-59/CHN/2015 CaKoV Chinese strain (GenBank No. MF062158). This kobuvirus genome has 8,184 nt, excluding the poly-(A) tail and contains a 7,335 bp ORF encoding a putative polyprotein precursor of 2,444 aa, a 601 bp 5′-UTR, and a 248 bp 3′-UTR (Fig. 3). Base content, 19.94% A, 20.95% G, 21.89% T, and 37.21% C (GC content 58.16%), is characteristic of the family Picornaviridae [14]. Strain SMCD-59 is 92.9%–96.6% identical based on nts (97.6%–98.1% identical based on aas) to four other CaKoV strains with available complete genome sequences (JQ11763/CH-1, KC161964/UK003, KF924623/12D049, and JN088541/PC0082). Aa identities of VP1, 3A, and 3B were 82.6%–87.9%, 81.9%–90.4%, and 76.9%–88.5%, respectively, with the other CaKoV strains. Results indicate that the VP1, 3A, and 3B genes have undergone greater evolutionary variation than previously reported (Table 3).

Graphical depiction of the complete SMCD-59/CHN/2015 CaKoV genome; predicted nucleotide cleavage site positions are shown
Phylogenetic analysis of our genome sequence with other published CaKoV genomes shows strain SMCD-59 to be more closely related to a Chinese (CH-1) and Korean (12D049) canine strain, than to other strains (Fig. 4A). The 3A gene phylogeny (Fig. 4B) places four canine strains (MF062158/SMCD-59, JQ11763/CH-1, KC161964/UK003, and KF924623/12D049) together, indicating a closer relationship among these strains from Asia and Europe, than to other, more distantly related strains from Africa and North America. Interestingly, phylogenetic analysis of the 3B gene shows all strains from Asia clustering together, separate from areas outside of Asia (Fig. 4C).

A neighbor-joining phylogenetic trees are shown based on the nucleotide sequences of strain SMCD-59/CHN/2015 CaKoV, together with those of other published CaKoV reference strains: specifically, the entire genome sequence (A), the 3A gene (B), and the 3B gene (C). The numbers at the nodes represent the bootstrap percentages supporting a particular branch occurring after 1,000 replicates. Isolates from this study are marked with black boxes
Discussion
CaKoV infection was identified in 50.46% of all the diarrheic fecal samples that we collected in southwest China, which is higher than other Chinese regions previously reported [10, 11]. This suggests that CaKoV has widely circulated among dogs in China. However, the pathogenic potential of CaKoV remains unclear. Moreover, previous studies have shown no significant difference in CaKoV prevalence between healthy and diarrheic dog samples [6]. Recently, it has been reported that CaKoV can cause systemic infections [13]. The pathogenicity of CaKoV needs to be studied in detail within the context of virus load in diarrheic versus healthy dogs, and in the role of co-infection with other enteroviruses.
Phylogenetic analysis of the 504 bp partial 3D gene sequence shows the 14 sequences in the study cluster into several different, loosely supported groups (Fig. 1). The CaKoV 3D sequences demonstrate considerable genetic diversity amongst themselves, as represented by horizontal branch lengths in the figure. This result is similar to a previous report in northeast China [10]. Interestingly, strain SMCQ-S6 clusters separately from the other strains, although bootstrap support for this distinction is not that strong (69%). Strain SMCQ-S6 has three nt replacements compared with the other 3D sequences in the study (positions 69, 111, and 189 in the alignment). Further targeted research is needed to determine the effect of these changes.
The VP1 protein of the Aichivirus is the most exposed and immunodominant portion of the kobuvirus capsid, and is the most variable structural protein in kobuviruses [14,15,16]. Furthermore, a proline-rich fragment of the Aichivirus VP1 protein (P228–P240) has been identified as a recognition motif for enteric receptor binding [17]. Regardless, detailed information regarding the CaKoV VP1 protein is quite limited.
The CaKoV VP1 aa sequences from our study have a similar proline-rich fragment at aa site P234–P244. Two aa substitutions occur in this region in these sequences, compared with other published sequences; whether this change affects function remains to be clarified. The five VP1 sequences from China that we generated and cluster together significantly (100% bootstrap), loosely associated with another Chinese sequence, and strongly separated from the other published VP1 sequences (98% bootstrap) in our phylogenetic analysis (Fig. 2). This suggests a unique evolutionary trend in the kobuviruses circulating among Chinese dogs, when compared with other countries.
We successfully obtained a complete CaKoV genome in this study. Sequence identity shows strain SMCD-59 to be more similar to both Asian and European strains than to North American strains. Phylogenetic analysis based on genome sequences shows CaKoV strains from Asia to be more closely related to each other than to European and American strains. Phylogenetic analyses of partial 3D and complete 3A genes are consistent with this result. Further studies will be required to ascertain whether the evolutionary relationships of any particular CaKoV gene can be used as an estimate of the overall evolutionary history of the various CaKoV strains.
Aa identities between the VP1, 3A, and 3B sequences of strain SMCD-59 and other strains ranges from 82.6%–87.9%, 81.9%–90.4%, and 76.9%–88.5%, respectively (Table 3). Until recently, the precise function of the 3A and 3B proteins were unclear for CaKoV. However, Klima et al. report that the 3A protein of the kobuvirus genus (i.e. Aichivirus A and Aichivirus B) act as a molecular harness to hijack host acyl-CoA-binding domain-containing protein-3 (ACBD3), which creates kobuvirus replication organelles at specific replication sites within the host cell [18]. Furthermore, the 3A protein of many picornaviruses plays an important role in viral replication [19]. Thus, we speculate that the 3A protein in CaKoV also participates in the replication of the virus. Therefore, further study should provide interesting results as to whether the 3A aa mutations we noted affect viral replication in CaKoV.
Conclusions
In conclusion, this study provides proof that CaKoV widely circulates in diarrheic dogs in southwest China, and shows considerable genetic diversity based on complete genomes, and on partial and entire gene sequences. Interestingly, the strains circulating in southwest China show unique evolutionary trends based on VP1 gene sequences. This may relate to the prominent role VP1 plays in host immune response. Furthermore, we successfully sequenced a complete genome from one of our samples. These findings contribute to our further understanding of epidemics and genetics, and expedite further study of CaKoV in China.
References
Khamrin P, Maneekarn N, Okitsu S, Ushijima H (2014) Epidemiology of human and animal kobuviruses. Virusease 25(2):195–200
Kapoor A, Simmonds P, Dubovi EJ, Qaisar N, Henriquez JA, Medina J, Shields S, Lipkin WI (2011) Characterization of a canine homolog of human aichivirus. J Virol 85(21):11520–11525
Li L, Pesavento PA, Shan T, Leutenegger CM, Wang C, Delwart E (2011) Viruses in diarrhoeic dogs include novel kobuviruses and sapoviruses. J Gen Virol 92:2534–2541
Carmona-Vicente N, Buesa J, Brown PA, Merga JY, Darby AC, Stavisky J, Sadler L, Gaskell RM, Dawson S, Radford AD (2013) Phylogeny and prevalence of kobuviruses in dogs and cats in the UK. Vet Microbiol 164(3–4):246–252
Di Martino B, Di Felice E, Ceci C, Di Profio F, Marsilio F (2013) Canine kobuviruses in diarrhoeic dogs in Italy. Vet Microbiol 166(1–2):246–249
Oem JK, Choi JW, Lee MH, Lee KK, Choi KS (2014) Canine kobuvirus infections in Korean dogs. Arch Virol 159(10):2751–2755
Choi S, Lim SI, Kim YK, Cho YY, Song JY, An DJ (2014) Phylogenetic analysis of astrovirus and kobuvirus in Korean dogs. J Vet Med Sci 76(3):1141–1145
Olarte-Castillo XA, Heeger F, Mazzoni CJ, Greenwood AD, Fyumagwa R, Moehlman PD, Hofer H, East ML (2015) Molecular characterization of canine kobuvirus in wild carnivores and the domestic dog in Africa. Virology 477:89–97
Soma T, Matsubayashi M, Sasai K (2016) Detection of kobuvirus RNA in Japanese domestic dogs. J Vet Med Sci 78(11):1731–1735
Li C, Wei S, Guo D, Wang Z, Geng Y, Wang E, Zhao X, Su M, Wang X, Sun D (2015) Prevalence and phylogenetic analysis of canine kobuviruses in diarrhoetic dogs in northeast China. J Vet Med Sci 78(1):7–11
Kong N, Zuo Y, Wang Z, Yu H, Zhou EM, Shan T, Tong G (2016) Molecular characterization of new described kobuvirus in dogs with diarrhea in China. Springerplus 5(1):2047
Di Martino B, Di Profio F, Melegari I, Robetto S, Di Felice E, Orusa R, Marsilio F (2014) Molecular evidence of kobuviruses in free-ranging red foxes (Vulpes vulpes). Arch Virol 159(7):1803–1806
Ribeiro J, Headley SA, Diniz JA, Pereira AH, Lorenzetti E, Alfieri AA, Alfieri AF (2017) Extra-intestinal detection of canine kobuvirus in a puppy from Southern Brazil. Arch Virol 162(3):867–872
Reuter G, Boros Ákos, Pankovics P (2011) Kobuviruses—a comprehensive review. Rev Med Virol 21(1):32–41
Yamashita T, Ito M, Kabashima Y, Tsuzuki H, Fujiura A, Sakae K (2003) Isolation and characterization of a new species of kobuvirus associated with cattle. J Gen Virol 84:3069–3077
Chen L, Zhu L, Zhou YC, Xu ZW, Guo WZ, Yang WY (2013) Molecular and phylogenetic analysis of the porcine kobuvirus VP1 region using infected pigs from Sichuan Province, China. Virol J 10:281
Zhu L, Wang X, Ren J, Kotecha A, Walter TS, Yuan S, Yamashita T, Tuthill TJ, Fry EE, Rao Z, Stuart DI (2016) Structure of human Aichi virus and implications for receptor binding. Nat Microbiol 1(11):16150
Klima M, Chalupska D, Różycki B, Humpolickova J, Rezabkova L, Silhan J, Baumlova A, Dubankova A, Boura E (2017) Kobuviral non-structural 3A proteins act as molecular harnesses to hijack the host ACBD3 protein. Structure 25(2):219–230
Fujita K, Krishnakumar SS, Franco D, Paul AV, London E, Wimmer E (2007) Membrane topography of the hydrophobic anchor sequence of poliovirus 3a and 3ab proteins and the functional effect of 3a/3ab membrane association upon RNA replication. Biochemistry 46(17):5185–5199
Acknowledgements
This work was funded by the 13th Five-Year Plan National Science and Technology Support Program (Grant number 2016YFD0500907). And, we also thank Steven M. Thompson, from Liwen Bianji, Edanz Editing China (http://www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling Editor: Diego G. Diel.
Rights and permissions
About this article

Cite this article
Li, M., Yan, N., Wang, M. et al. Prevalence and genomic characteristics of canine kobuvirus in southwest China. Arch Virol 163, 459–466 (2018). https://doi.org/10.1007/s00705-017-3648-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-017-3648-y